
Tert-butyl phenols are important commodity chemicals, with annual production exceeding 120,000 t per annum. They have multiple industrial applications including agrochemicals, antioxidants- in particular 2,6-di-tert-butyl-4- methylphenol (BHT, Figure 1), foods, flavours and fragrances and pharmaceuticals.1 BHT in particular, used as an antioxidant in polymers and oils and prepared by reaction of p-cresol with isobutylene in the presence of an acid exchange resin at elevated pressure, accounts for the vast majority of the global production of tert-butylated phenol substrates.2a A range of materials catalyse the tert-butylation of simple aryl phenols including sulfuric and phosphoric acids, boron trifluoride, activated clays and zeolites as well as the strongly acidic ion exchange resins eluded to above.2b A good review by Pettus in Synlett last year summarises these methods very nicely.3 Most of the methods generally produce mixtures of regioisomers that are separated on industrial scale by fractional distillation. Often thermal isomerisation to the most thermodynamically stable molecule occurs during the purification process.4
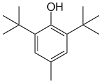
Using an alky group as a positional protecting group for aromatic compounds is not something I’d come across before. The bulky tert-butyl group has been used to block the reactive ortho and/or para position of phenols, taming their reactivity and enabling control of regioselectivity in down-stream chemistry.5 Deprotection via a retro-Friedel Crafts process regenerates the phenol.6 Figure 2 shows a simple example: Friedel Craft tert-butylation and protection of the 4-position of anisole, zinc chloride catalysed Friedel Craft ortho– acetylation and retro-Friedel Craft tert-butyl cleavage to unmask the para position.7 I guess zinc is not sufficiently Lewis acidic to cleave the tert-butyl group in the second step.
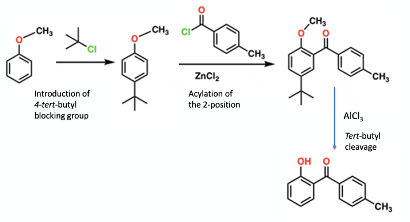
The reason that this chemistry is of interest to me is that one of the case studies we use in our Chemical Development & Scale-Up course uses this tert-butyl protecting group approach, and an unexcepted scale-up effect results in poor product yield and formation of an impurity not observed during lab-scale development work (Figure 3). The chemistry was developed by Smith Kline & French in the 1980’s towards a synthesis of a L-94901, a thyroid hormone mimetic.8
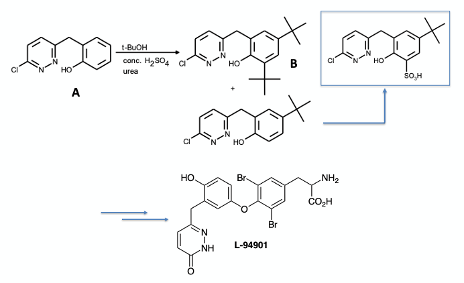
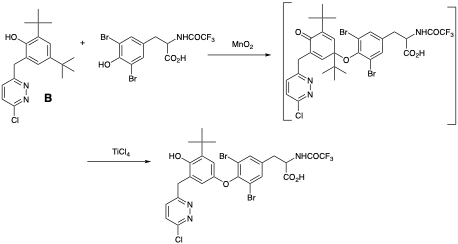
L-94901 is assembled via an oxidative coupling of two phenols using MnO2, followed by Lewis acid catalysed de-tert-butylation of the unstable quinol ether using TiCl4 (Figure 4)8a These type of oxidative dimerisation processes are known to generate impurities in process using THF stabilised BHT as reaction solvent.8b
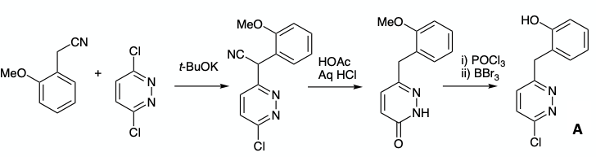
The phenol precursor (A) was prepared by base-catalysed alkylation of 3,6-dichloro- pyridazine with 2-methoxybenzyl cyanide (t-BuOK/t-BuOH) to generate a nitrile intermediate that was hydrolysed in glacial acetic acid to give the pyridazione. Chlorination with POCl3 and demethylation gave phenol A (Figure 5) .9
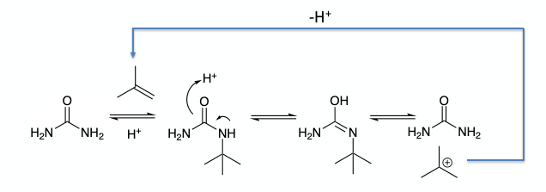
And so to the crux of the problem-converting phenol (A) to the di-tert-butyl protected intermediate (B) and the issues with scaling the chemistry (Figure 2). In the lab this reaction worked very well. The tert-butyl groups function as positional protectors of the phenolic ring so that manipulation of the heterocyclic ring can safely be carried out in the next stage of the synthesis. They are easily removed by a reverse Friedel Crafts reaction with AlCl3 in toluene.10 Monoalkylation of phenol (A) was rapid using conc. H2SO4-urea-tert-BuOH/H2O. Other alkylation conditions were tried, such as isobutene/c. H2SO4, t-butyl acetate- c. H2SO4 / H2O, t-butyl bromide- c. H2SO4 / H2O, t-butyl alcohol- c. H2SO4 / H2O, however these methods did not offer any advantages over the tert-butyl alcohol-urea system. The urea is used in large excess in the reaction (20 equivalents). Tert-butanol is dehydrated in the acidic reaction mixture, generating isobutene (the tertiary carbocation precursor), and reaction with urea- the major nucleophilic species present in the reaction mixture- generates a mixture of non-volatile mono– and/or bis– N-tert-butyl intermediates.11 Isobutene itself boils at -6.9°C and is lost to a large extent (in a non-pressurized systems) though off-gassing. Figure 6 shows the general idea. The lack of volatility of the N-alkyl intermediate ensures a constant equilibrium supply of tert-butyl carbocation- some of which is trapped by the phenol substrate and some converted back to isobutene. The urea system gave better results than tert-butanol/H2SO4 alone. Generally, these uncatalyzed reactions need to be heated and give uncontrollable off-gassing.12
In the case of our phenol (A), mono-tert-butylation in the para-position was rapid, however the second ortho-alkylation was a much slower process (presumably due to steric effects), requiring large excesses of tert-butanol and urea. In the plant, the second reaction was even slower and a sulphonic acid by-product, formed from competing reaction of the mono-tert-butyl product with sulphuric acid, was produced as a major impurity (Figure 2).13
Preventing this side-reaction by, for example, changing the acid strength or using a different acid altogether could possibly solve the problem. However, the main issue was that the isobutylene was being lost from the vessel, starving the reaction of alkene. In the plant the exit gasses were scrubbed via a condensation process that created a slight vacuum in the system. This further exacerbated the problem. For safety reasons the reaction vessel was inerted using a slow nitrogen gas flow that also carried away the isobutene. The problem was solved by careful consideration of these issues and further detailed optimisation.
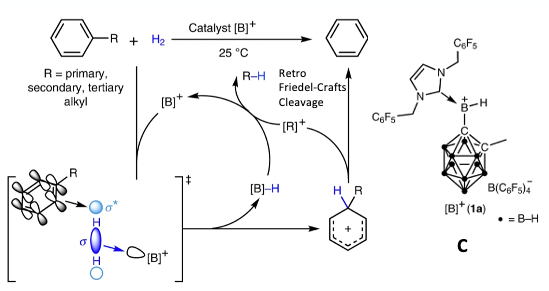
Finally, before I close, an interesting paper recently published by Wang et al in Nature Catalysis describes the application of borenium complex (Figure 7, C) to catalyse the selective hydrogenolysis of unstrained C(aryl)– C(alkyl) bonds in alkylarenes in the absence of a directing group.14 These type of reactions are run industrially but require forcing conditions: typically using a metal catalyst at temperatures above 300 °C with high pressures of hydrogen gas (40–200 bar).
The borenium catalyst (C) has been used in the past for methane activation.15 The described retro-Friedel Crafts reaction works at ambient temperature and was inspired by the frustrated Lewis pair activation of hydrogen (Figure 7). The arene is believed to act as a π-electron donor, activating dihydrogen (in the presence of the Lewis acid), leading to the formation of a hydride species and a Wheland complex (Figure 7). A retro-Friedel–Crafts reaction of the Wheland complex gives the arene and a carbenium intermediate.
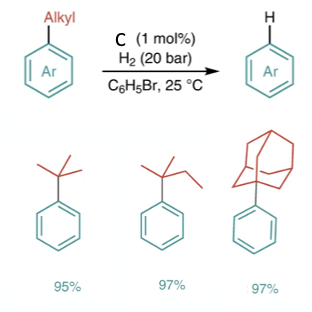
The reported reaction conditions are straightforward: alkylarenes (1.0 mmol) and borenium catalyst (C, 0.010 mmol) in bromobenzene (0.5 ml) are stirred under hydrogen gas (20 bar) at room temperature for 24hrs. The reported yields are mostly by GC (something I don’t really like in academic papers). Figure 8 shows a few tert-alkyl substrates reported in the paper. The scope, in terms of aryl substitution, seems somewhat limited at present (Me, Cl, Br, F). Phenols are not described.
The above case study is a good example of the importance of running small-scale “stressing” experiments during reaction development that closely mimic the extended addition and processing times encountered when running a large-scale process. It can be difficult to anticipate these issues and early scale-up is still very much a learning exercise.
For more information on our Chemical Development and Scale-Up course please see our website or contact me directly.
See you next time.
References:
- Phenol derivatives: H. Fiege (Bayer) et al, Ullmans encyclopaedia of Industrial chemistry 2012, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim DOI: 10.1002/14356007.a19_313
- a) Butylated hydroxytoluene analogs: synthesis and evaluation of their multipotent antioxidant activities: A. Ariffin et al, Molecules 2012, 17, 7645-7665; alkylation of phenol with tertiary butyl alcohol over zeolites: N. Pradhan et al, Org. Process Res. Dev. 2002, 6, 132-137
- Strategies for ortho–tert-butylation of phenols and their analogues: T. Pettus et al, Synlett 2022, 33, 575-580; Total synthesis of (±)-Quadrangularin A: Z. Hou et al, Angew. Chem. Int. Ed. 2006, 45, 7609-7611
- Process for the alkylation of aryl hydroxides: DE1443346A1 (Union Carbide)
- A preparative route to alkoxyphenols using t-butyl function as a positional protective group: M. Tashiro et al, Org. Prep. Pres. Int. 1984, 16, 155-16; Selective synthesis of aromatic compounds using positional protecting groups: Tashiro et al, Org. Prep. Proc. Int. 1984, 16, 155-164; Synthesis 1979, 921-936
- A facile and mild de-tert– butylation reaction: N. Lewis et al, Synth. Commun. 1988, 18, 1783-1793; De-tert-butylation of substituted arenes: H. Tashtoush et al, Tetrahedron 1998, 54, 14157-14177l,
- Tert-butyl as a blocking group in the synthesis of ortho-hydroxyphenones: M. Kulka, Am. Chem. Soc.1954, 76, 5469-5471
- a) Synthesis of thyroid hormone analogues. Part 2.- oxidative coupling approach to SK&F L-94901: D. Hickey et al, J. Chem. Soc. Perkin Trans. 1, 1988, 3097-3102; b) Process research and development of a dihydropyrimidine dehydrogenase inactivator: large-scale production of Eniluracil using a Sonagashira coupling: J. Cooke et al, Org. Process Res. Dev. 2001, 5, 383-386
- Synthesis of thyroid hormone analogues. part 1. preparation of 3’- heteroarylmethyl-3,5-di-iodo-l-thyronines via phenol-di nitrophenol condensation and relationships between structure and selective thyromimetic activity: P. Leeson et al, Chem. Soc. Perkin Trans. 1, 1988, 3085-3096; Synthesis of thyroid hormone analogues. Part 3. Iodonium salt approaches to SK&F L-94901: D. Hickey et al, J. Chem. Soc. Perkin Trans. 1, 1988, 3103-3111
- De-tert-butylation of substituted arenes: H. Tashtoush et al, Tetrahedron 1998, 54, 14157-14177; Method for removing tert-butyl groups from tert-butyl phenol compounds: EP1151986B1 (Honshu chemical industry)
- Use of urea and its N- substituted tert-butyl derivatives in tert-butylation of aromatic compounds: N. Nevrekar et al, Chem. Ind. 7 March 1983, 206-207
- Stabilization of the hindered urea bond through de-tert-butylation: J. Cheng et al, Chem. Commun. 2021, 57, 3812-3815
- Studly of phenol sulfonation by concentrated sulfuric acid: kinetics and process optimisation: J. F. Chen et al, Chem. Eng. Sci. 2019, 202, 15-25
- Boron-catalysed hydrogenolysis of unactivated C(aryl)-C(alkyl) bonds: H. Wang et al, Nat. Catal. 2023, 6, 16-22
- Cationic tricoordinate boron intermediates: borenium chemistry from the organic perspective: E. Vedejs et al, Chem. Rev. 2012, 112, 4246-4282; Methane activation by a borenium complex: H. Wang et al, Chem. 2021, 7, 1843-1851




