
1. Using an approved drug as a diazomethane surrogate1
A recent paper published in Angewandte Chemie describes the use of Temozolomide (TMZ) as a solid weighable diazomethane surrogate.
TMZ is an approved drug and is the standout treatment for gliobastoma. It is stable weighable solid, with reasonable water solubility. Under physiological conditions , pH 7.4, 37°C, it hydrolyses with loss of CO2 to form MTIC which generates methyldiazoium- a reactive intermediate that methylates DNA in tumour cells (Scheme 1).
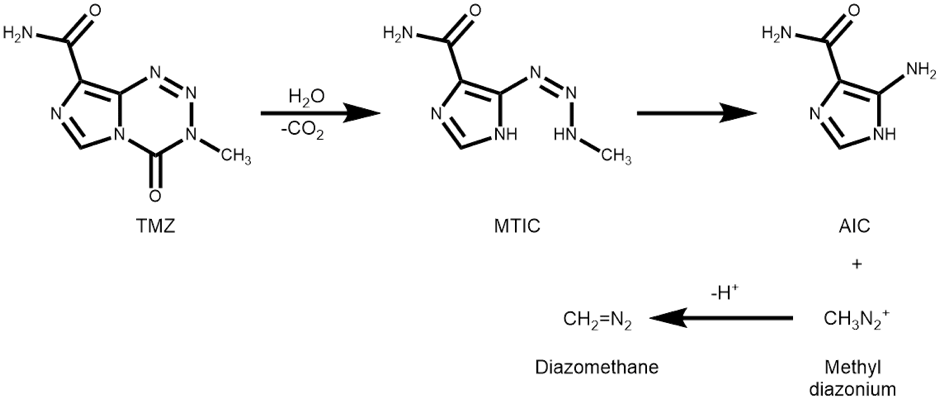
Taking a carboxylic acid and treating it with 2 equivalents of TMZ and 2 equivalents of Na2CO3in dioxane/water (9:1) at 60°C results in esterification. In some cases a second dose of TMZ and Na2CO3is required.
Yields are typically 70-95% and unusually the 26 examples include some complex drug molecules (e.g. indomethacin, sulindac, telmisartan) and natural products (e.g. gibberellic acid, retinoic acid) as substrates.
The reaction is very selective. Phenols do not react, MOM, THP, Boc and FMOC protecting groups survive intact.
Other analogues of TMZ can be made and examples of making ethyl esters and CD3 esters are also described.
In addition, the diazomethane generated from TMZ can be used to cyclopropanate styrenes. In this case the reaction is carried out in 6M KOH with and iron porphyrin catalyst at room temperature. Yields are 55-93% for liquid styrenes, but lower for solid styrenes where toluene has to be added as a cosolvent.
The one drawback to the use of TMZ for in situ diazomethane generation is the atom economy and the number of equivalents of TMZ required, but as a lab method it is considerably safer than traditional methods of generating diazomethane.
2. Selective cross-coupling with polyhalogenated aryl fluorosulfates2
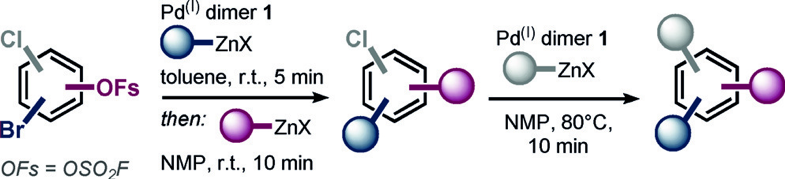
Cross coupling reactions are very commonly used in a variety of industries such as pharmaceutical, agrochemical and materials. This work initially is about selective coupling with aryl bromides in the presence of fluorosulfates, and then a second coupling with the fluorosulfate (FSO) acting as a cheaper and more atom efficient alternative to the triflate or nonaflate group. Selective sp2-sp2and sp2-sp3 coupling at the bromide is takes place rapidly and selectively at room temperature with the Pd(I) dimer (t-Bu3PPdI)2 as catalyst with yields in the range 66-94%. The reactions are run in toluene-THF and are complete in 5 minutes at room temperature with 2.5 mol % catalyst. The selectivity for reaction at the bromide seems to be independent of steric effects or the relative position of the 2 groups.
Chloro-fluorosulfates on the other hand react exclusively with organozinc species at the FSO position. In this case a more polar solvent mixture, NMP-THF, is required for the less reactive FSO group.
Starting in toluene and then adding NMP as solvent allows a 2- or 3-component sequence to be carried out where a bromochlorofluorosulfate is first substituted at the bromide, then at the FSO and finally (for the 3-component sequence) at the chloride position (Scheme 1).
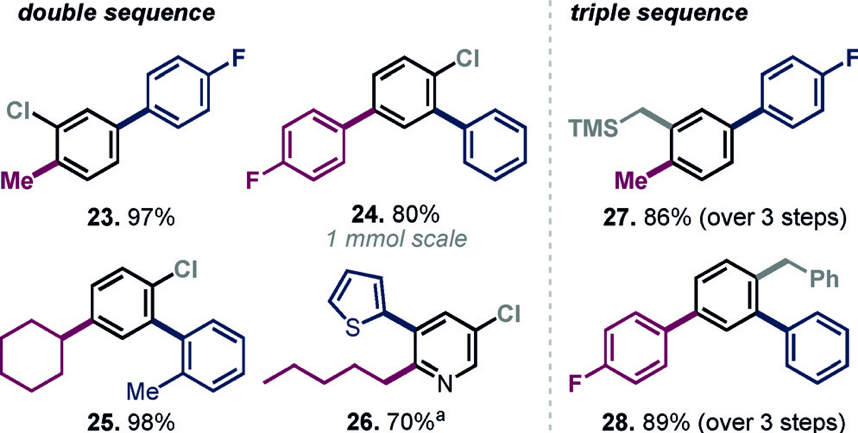
The yields for both a double and triple sequence are impressive.
- R.L. Svec and P.J. Hergenrother, Angew. Chem. Int. Ed., 2020, 59, 1857-1862.
- Reference: Mendel, I. Kalvet, D. Hupperich, G. Magnin and F. Schoenbeck, Angew. Chem. Int. Ed., 2020, 59, 2115-2119.




