
As synthetic chemists we are very familiar with making chemical bonds, particularly C-C bonds, and building complex molecules. Having said that, fundamentally, with sufficient energy and entropy in the driving seat, it should be quicker and easier to break molecules apart. Deconstructive processes are not, however, as common as one might think- particularly cleavage of C(sp3)-C(sp3) bonds.1 In fact the carbon-carbon single bond is arguably one of the least reactive ‘functional groups’ in chemistry. There are of course brute force methods employed by the petrochemical industry- very high temperature catalytic cracking. These methods are suitable for inert hydrocarbons but would obliterate a sensitive molecule or labile functional group. By comparison carbon-carbon double bond cleavage reactions and functionalization are well known to organic chemists and utilized industrially- for example olefin metathesis (viaalkene scission and regeneration of C=C bonds)2 and ozonolysis (forming C-O bonds).3 In addition stochiometric non-metallic oxidants such as NaIO4, H5IO6 or transition metal based reagents, such as Pb(OAc)4and KMnO4, are commonly used for oxidative cleavage of C-C s-bonds. These reagents can be somewhat over-zealous in their reactivity, precluding sensitive substrates and functional groups. Recently Sarpong et alhas described a deconstructive functionalization process for converting saturated nitrogen heterocycles such as piperidines or pyrrolidines into halogen-containing acyclic amine derivatives through sequential C(sp3)–N and C(sp3)–C(sp3) single-bond cleavage followed by C(sp3)–halogen bond formation,4 including fluorination (Scheme 1).5
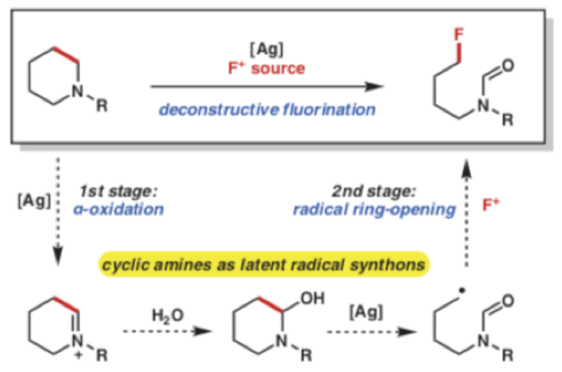
Scheme 1: Deconstructive functionalization of piperidine
Early work on transition metal mediated catalytic cleavage of a C-C single bond using a soluble rhodium complex was reported by David Milstein in 1998.6 Reaction of [RhCl(C8H14)2]2with a mesitylene derivative containing an appropriately substituted diphosphine under mild hydrogen pressure (25psi) resulted in selective cleavage of the strong C-C bond (Scheme 2). Triethyoxysilane could also be used as terminal reductant.
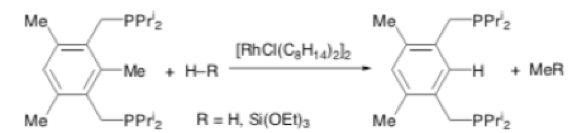
Scheme 2: Rh catalysed Me group cleavage
Nature has evolved metalloenzymes based on iron or copper for metabolism of steroids, amino acids and xenobiotics (including drug molecules).7 These enzymes are dioxygenasesthat use diatomic oxygen in the key steps of their metabolic processing.8 P450 enzymes have been used synthetically to N-dealkylate and ring open saturated nitrogen containing heterocycles.
An interesting example of biochemical C-C bond cleavage is the metabolic activation of the anti-inflammatory prodrug nabumetone. The molecule undergoes P450 mediated hepatic oxidative cleavage of the aliphatic side chain to generate the active carboxylic acid (6-MNA) (Scheme 3).9
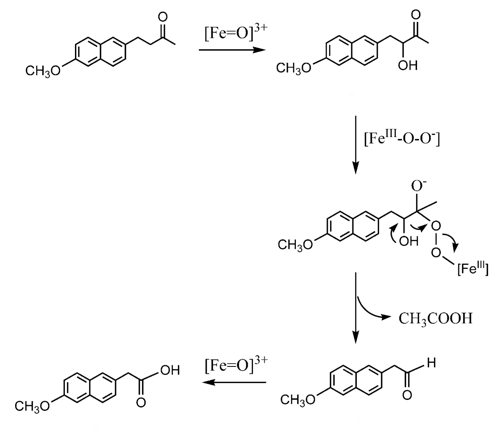
Scheme 3: P450-catalysed oxidation of nabumetone to 6-MNA
The attraction of these deconstructive processes is that they have potential utility in functionalization of active molecules and could also be used to construct small molecule building blocks and feedstocks. A couple of recent papers focusing on carbon-carbon bond cleaving reactions demonstrate how useful these approaches can be. The first is a publication by the Beller group at the Leibniz institute on copper-mediated cleavage of C(sp3)-C(sp3) bonds in amines (Angew. Chem. Int. Ed.2019, 58, 10693-10697) and the second an intriguing ruthenium-catalysed reductive cleavage of unstrained aryl-aryl bonds- a kind of retro-Suzuki process- by Dong et al, at the University of Chicago (J. Am. Chem. Soc.2019, 141, 18630-18640).
The work by Beller et al began by studying C-H oxidation ofn-tributylamine, screening a range of metals, oxidants ligands and solvents. They quickly identified copper, using air as the oxidant. After several rounds of optimization, the preferred conditions were identified (5 mol % CuCl, 2 eqv. pyridine 30 bar air, 100 °C in acetonitrile). Dibutylformamide was obtained in 90% yield with no apparent sensitivity of the reaction to water. A wide range of tertiary aliphatic amines are exemplified in the paper with good chemo- and regioselectivity (Scheme 4).
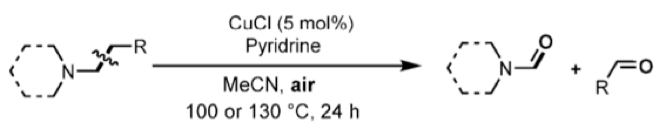
Scheme 4: C-C bond cleavage in tertiary amines
Derivatization of morpholine and piperazine, which constitute a large number of drugs and drug-like molecules, was also possible under slightly modified conditions (3 mol % Cu(CF3SO3)2, 20 mol % pyridine, MeCN, 100 °C, 24 hours). Late stage C-C cleavage of drugs such as the antimicrobial agent Linezolid and the NSAID Emorfazone and natural products such as Citronellal was also successful (Figure 1).
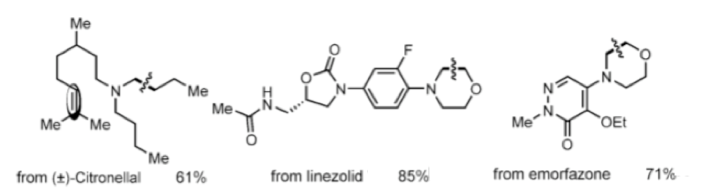
Figure 1: Late stage C-C bond cleavage
Beller’s team undertook a number of experiments to define the mechanism of the process. Since addition of pyridine significantly improves the reaction conversion, the stability of the additive was confirmed by using [D5]-pyridine. No deuterium exchange was observed using Gc/Ms analysis. Subjecting 4-phenylmorpholin-3-one and 4-phenylmorpholin-2,3-dione to the standard reaction conditions did not generate product excluding them as putative intermediates.
Radical trapping reagents however, including TEMPO and BHT, stopped the reaction suggesting radical intermediates were important in the process. Air was also required in combination with the copper catalyst. Water was confirmed not to be an oxygen donor by 18O labelling studies.
EPR and UV/Vis studies confirmed a rapid disappearance of the CuII EPR signal on addition of N-phenylmorpholine and formation of a temporary signalwith unresolved hyperfine structure typical of an organic radical cation. The activation of oxygen by the CuIor the radical cation to form a superoxide species was not elucidated and is likely to be a complex process. Reduction of CuII to CuIwas followed also by UV/Vis measurements of ligand to metal charge transfer (Scheme 5).
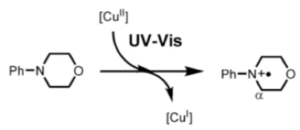
Scheme 5: Formation of a radical cation and UV/Vis detection of CuII to CuI transition
The second highlighted paper concerns cleavage of an unstrained non-polar C(aryl)-C(aryl) bonds- one of the toughest C-C bonds to break. The JACS paper by Dong et aldescribes development of a ruthenium catalysed reductive cleavage of bonds of this type.10 The latest work builds on their earlier publication describing the catalytic activation of unstrained C(aryl)−C(aryl) bonds in 2,2’-biphenols.11
In this modification the biaryl substrates require a suitable directing group (DG) in the 2, 2’ positions- and encompasses heterocycles such as pyridine, quinoline, pyrimidine and pyrazoles (Scheme 6). In addition to hydrogen gas as the terminal reductant, reagents such as Hantzsch esters, silanes and alcohols can also be used.
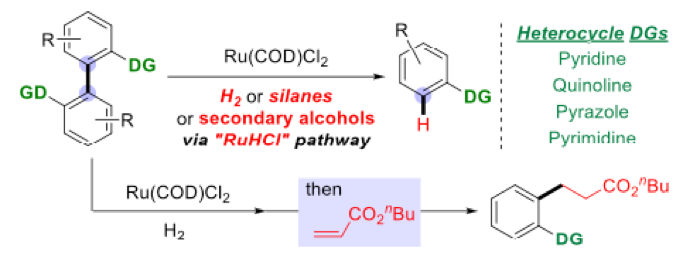
Scheme 6: Cleavage of unstrained aryl C-C bonds with Ru
An interesting extension of the process was the one-pot C-C bond activation/C-C bond formation approach. This relies on the ruthenium catalyst remaining active after the initial bond cleaving reaction. Addition of acrylate enabled selective ortho– alkylation of the C-C cleavage product in high yield (Schemes 6 and 7). Similar utilization of remaining active catalyst in subsequent (or alternative) transformations has been reported for metathesis, coupling and reduction chemistries.12
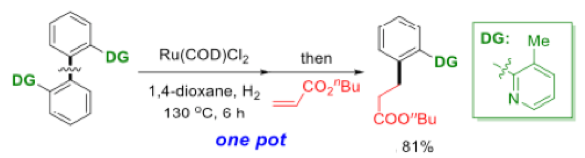
Scheme 7: One pot C-C activation and C-C bond formation
Establishing the reaction mechanism was aided by obtaining an X-ray structure of the resting state of the catalyst (Figure 2). This showed an η4-coordinated ruthenium(II) complex. In addition, DFT calculations and experiments showed involvement of a ruthenium(II) monohydride-mediated C(aryl)−C(aryl) activation step.
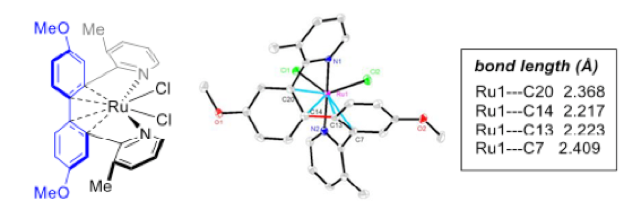
Figure 2: X-ray structure of the catalyst resting state
These papers both show that C-C bond cleavage reactions have the potential to be a useful strategic tactic in retrosynthetic analysis. Breaking up just got that little bit easier…
References:
- Recent advances in transition-metal catalysed functionalization of unstrained carbon-carbon bonds: F. Chen et al, Rev. 2014, 114, 8613 – 866; Cleavage of Carbon—Carbon Single Bonds by Transition Metals: M. Murakami et al, Top. Organomet. Chem.1999, 3, 97.
- Ruthenium-based heterocyclic carbene-coordinated olefin metathesis catalysts: R.Grubbs et al, Chem Rev. 2010, 110, 1746-1787.
- Alkene ozonolysis: P. Dussault et al, Tetrahedron 2017, 73, 4233-4258.
- Deconstructive diversification of cyclic amines: R. Sarpong et al, Nature 2018, 564, 244-248.
- Deconstructive fluorination of cyclic amines by carbon-carbon cleavage: R. Sarpong et al, Science, 2018, 361, 171-174.
- Catalytic selective cleavage of a strong C-C single bond by rhodium in solution: D. Milstein et al, Chem. Commun. 1998, 687-688.
- Formation and Cleavage of C−C Bonds by Enzymatic Oxidation− Reduction Reactions: F. Guengerich et al, Chem. Rev. 2018, 118, 6573-6655.
- The Mechanism of Formation ofN-Formylkynurenine by Heme Dioxygenases: E. Raven et al, J. Am. Chem. Soc. 2011, 133, 16251-16257.
- Carbon-carbon bond cleavage in activation of the prodrug Nambumetone: F. Varfaj et al, Drug Met. Disp. 2014, 42, 828-838.
- Ruthenium-catalyzed reductive clevage of unsaturated aryl-aryl bonds: reaction development and mechanistic studies: G. Dong et al, J. Am. Chem. Soc. 2019, 141, 18630-18640.
- Catalytic activation of unstrained C(aryl)−C(aryl) bonds in 2,2’-biphenols: Dong et al, Nat. Chem. 2019, 11, 45-51.
- Catalyst farming: Reaction rotation extends catalyst performance: A Elhage et al, chem. Sci. 2019, 10, 1419-1425.




