
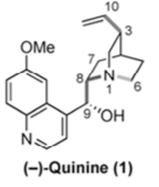
The naturally occurring cinchona alkaloids, and Quinine in particular, have been of interest to synthetic chemistry practitioners for many years, due ostensibly to its antimalarial activity, particularly against resistant strains of the disease. The original synthesis, by Woodward and Doering in 1944 was heralded as a milestone in the art of synthetic organic chemistry,[1] and elegant syntheses by other renowned chemists including Stork, Kobayahi, Jacobsen and Aggarwal have appeared in the literature over the years.[2]
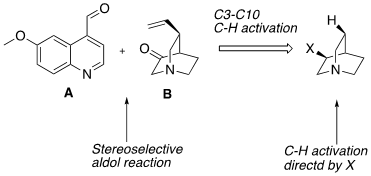
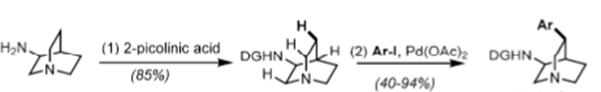
In order to structurally modify a natural product and enhance its activity or attenuate its off-target pharmacology a concise, robust synthesis is required. A resect paper by Maulide etal (Angew. Chem. Int. Ed. 2018, 57, 17037) describes a short, flexible approach to both the natural (-) and unnatural (+) quinine and several C3 aryl derivatives from 3-aminoquinuclidine. Their approach involved two stereoselective synthetic steps, namely a selective C(SP3)-H activation step and a novel C8-C9 disconnection through an aldol reaction used to combine two key advanced intermediates (see above).[3] A C7 amino substituent (quinuclidine numbering) was modified to produce a picolinamide directing group which directed clean arylation using Pd catalysis with complete regio- and diastereocontrol for a range of substituted aryl groups (Scheme 1). Direct vinylation at this position, however, was unsuccessful.
(C)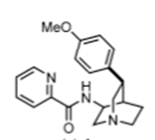
Scheme 1: picolinamide directed CH- activation
The required vinyl- intermediate (B) was prepared from the 4-methyoxyphenyl derivative (C) in a 4-step sequence involving RuCl3 catalysed oxidative degradation to the carboxylic acid, conversion to the Weinreb amide, DIBAL-H reduction to a masked aldehyde and Wittig olefination. Zn/HCl was used to remove the directing group and conversion of the free amine to ketone achieved with IBX. Conversion of the anisole to a carboxylic acid is an interesting disconnection originally reported by Caputo and greatly improved by Sharpless.[4]
Final aldol coupling of (B) with aldehyde (A) was achieved using LiHMDS at low temperature, but due to rapid epimerisation at C8 an in situ derivatization sequence was developed involving addition of Ti(OiPr)3Cl followed by conversion to a sulfonyl hydrazone (Scheme 2). The required hydrazone was prepared in 76% yield and >16:1 d.r (Me hydrazone derivative).
The final step in a synthesis should be a pleasant experience but proved to be a significant challenge for the team. Reduction of the tosyl hydrazone proved all but impossible, however switching to the less sterically encumbered mesyl hydrazone and use of LiALH4 in methanol (trimethoxyaluminium hydride) gave the natural product in 53% yield.
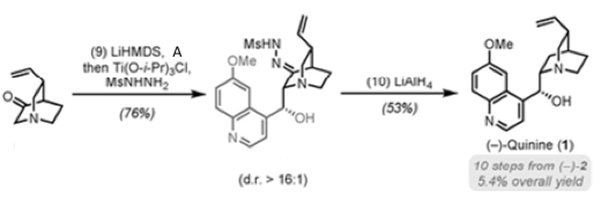
Scheme 2: end game synthesis: selective aldol and hydrazone reduction
(-) Quinine was prepared in 5.4% overall yield in 10 synthetic steps from (-)-3-aminoquinuclidene. C3 Aryl derivatives of quinine show enhanced antimalarial activity in vitro and in vivo and the authors suggest potential therapeutic application of these derivatives.
[1] J. Am. Chem. Soc. 1944, 66(5), 849; ibid 1945, 67, 860
[2] Angew. Chem. Int. Ed. 2018, 57, 17037, references 3-5
[3] Use of the picolinamide directing group for C-H activation: Angew. Chem. Int. Ed. 2011, 50, 5192
[4] Tett. Lett. 1981, 22, 3815, J. Org. Chem 1981, 46, 3936.




