
A couple of interesting papers published recently on aryl iodination. I sure I don’t need to reiterate how useful iodinated intermediates are in synthesis- most significantly their heightened reactivity in metal catalysed cross coupling chemistry,1 and, more recently their application as organocatalysts in hypervalent iodine chemistry.2 As is common in synthetic science, the intermediates you really need are the ones that are most difficult to make and iodoarenes are no exception. They are generally more difficult to access than the corresponding chlorides or bromides due largely to the limited availability of electrophilic iodinating reagents of comparable reactivity to their lighter halogen cousins. Elemental iodine (I2) and electrophilic iodinating reagents such as N-iodosuccinimide (NIS) and 1,3-diiodo-5,5-dimethylhydantoin (DIH) are generally not reactive enough to iodinate electron-poor aromatic or heteroaromatic systems, and if they react at all the yields are low and the reactions non-selective.
Tobias Ritter has succeeded in accessing previously unexplored electrophilic iodinating agents- sulfonyl hypoiodites (although they do claim the intermediate is putative).3 Attempts to make the methanesulfonate reagent by reaction of iodide with bis(methanesulfonyl) peroxide were thwarted by the fact that the peroxide precursor is explosive. Thankfully for those involved in the project (and anyone who subsequently implements the chemistry) an alternative in situ method was developed using silver mesylate and molecular iodine (Figure 1).
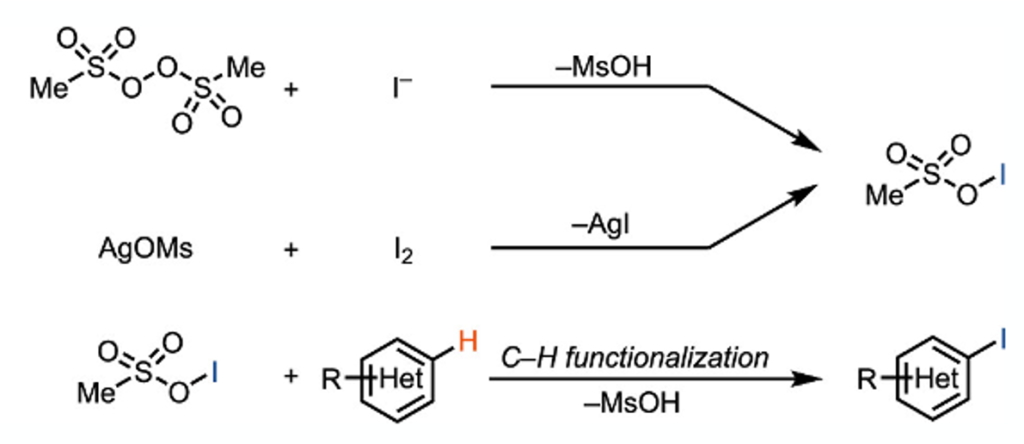
The in situ generation from the silver salt gave comparable results to the peroxide mode of formation. In addition to the obvious safety benefits, the silver salt approach could be used to generate a range of hypoiodites with different reactivity depending on the nature of the sulfonate. Figure 2 shows a comparison of the sulfonyl hypoiodite with other iodinating reagents using tetrahydrobenzo furanone as a test substrate.
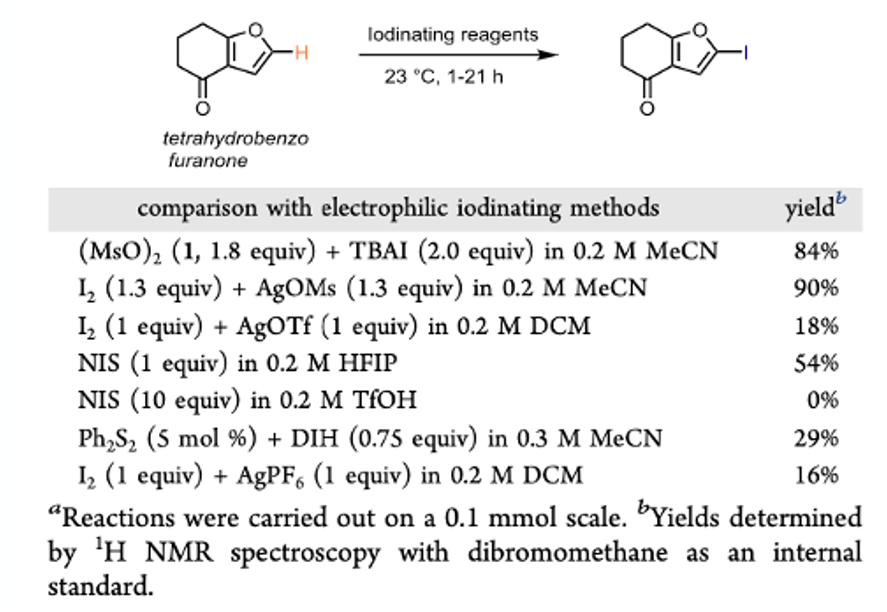
Really the only reagent that comes close to the hypoiodite in terms of yield is NIS in the weaky acidic solvent hexafluoroisopropanol (HFIP). However, the effectiveness of NIS drops off dramatically with electron poor substrates or those with functionality-derived complexity.
Simple arenes worked well using this process, generally with high selectivity, however nitrobenzene and aniline both failed to iodinate-presumably due to facile side-reactions. N-heterocycles- pyridines, pyrazoles and pyrroles- substituted with carboxylic acids, esters, amines, sulfonamides, and phthalimide substituents were all well tolerated. Silver mesylates, tosylates or triflates were used as precursors in the various examples described.
A (carefully selected) range of small-molecule drugs with additional structural complexity worked well with good positional selectivity (Figure 3). The strychnine example is particularly interesting, with complete selectivity for the 4-position of the aromatic ring observed with no reported iodination of the alkene or reaction at the tertiary nitrogen.
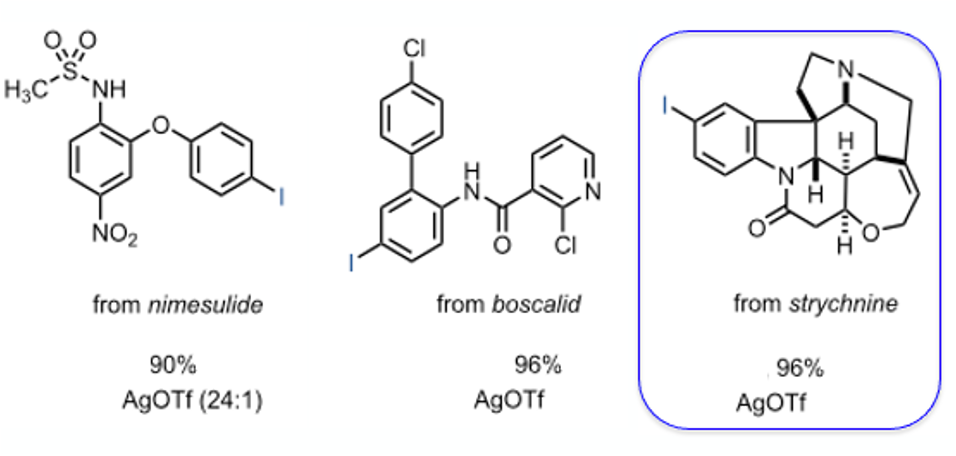
The hypoiodites looks like an interesting reagents and the chemistry appears relatively straightforward to run. Exercise caution with any materials containing a halogen-oxygen bond. They can be somewhat energetic so any scale-up work should be undertaken with all the usual diligence and safety testing.
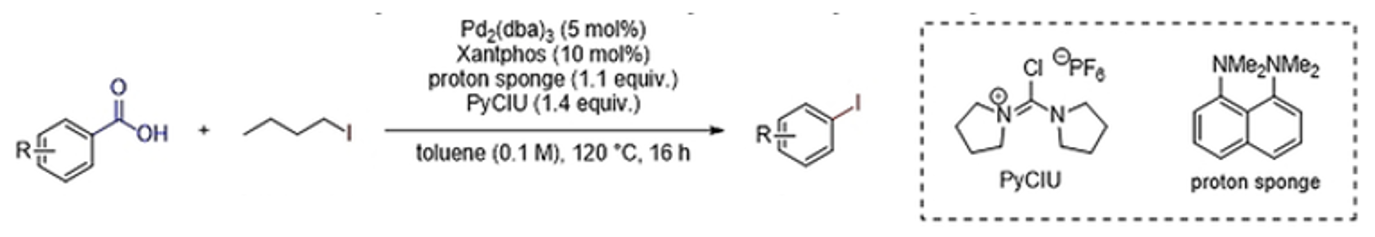
The second approach is work published by Bill Morandi,4 and is attractive because it utilises benzoic acids as starting materials. These precursors are generally cheap, readily available (in a wide range of substitution patterns) and are regarded as feedstock chemicals. In essence, the paper describes the development of a reaction for direct conversion of aryl carboxylic acids into aryl iodides (Figure 4).
This is not a new process as such- the Hunsdiecker reaction, in which silver carboxylate salts are converted to halides, has been known since the 1930’s. This older process does have its limitations however, including the use of stoichiometric metal and the need for preactivation of the acid. Photocatalytic methods using iridium catalysts have also been developed, but again are not straightforward to execute, particularly at any kind of scale.5
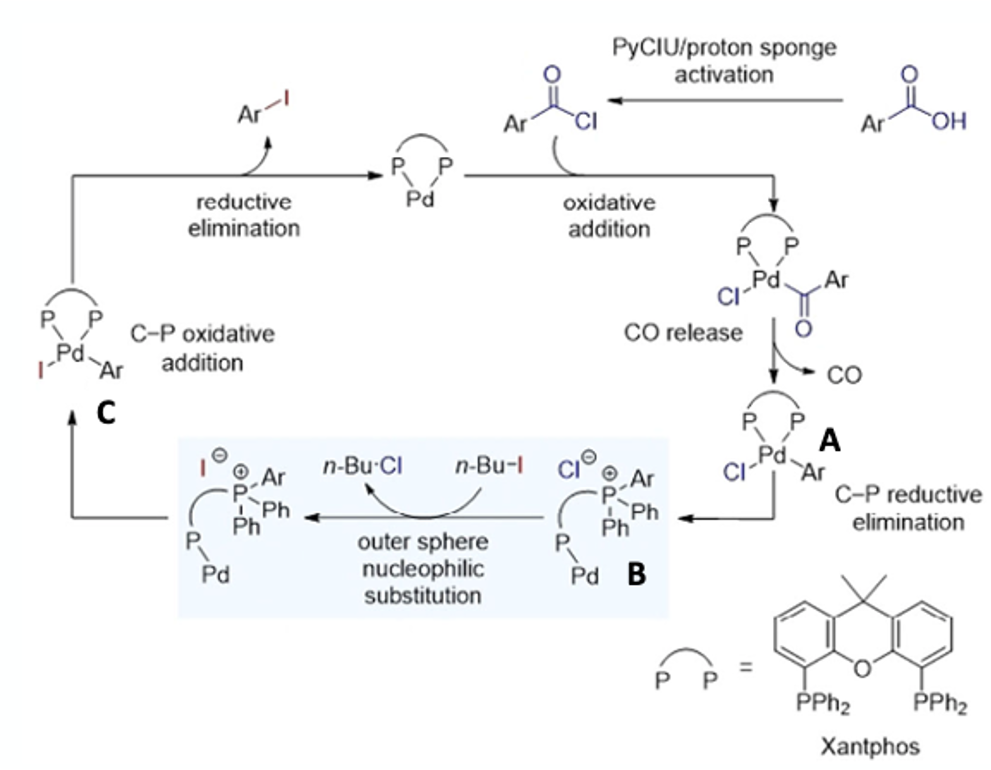
In 2018 Morandi published a single-bond metathesis reaction for the synthesis of aryl iodides from acid chlorides using Pd(0)/Xantphos.6 Although interesting, preactivation of the acid to acid chloride was required and, of course, the stoichiometric by-product is also an acid chloride- making purification extremely difficult. Having demonstrated that the Pd/Xantphos catalyst system could facilitate reductive elimination of C-I bonds, the team have extended this methodology using in situ generation of the acid chloride and conversion to the iodoarene using an iodide source. In this case the by-products are CO gas and a volatile chloroalkane (n-butyl chloride boils at 78°C). Following some initial screening of iodide sources, alkyl iodides proved the most suitable. Ultimately a combination of Pd2(dba)3 (5 mol%), Xantphos (10 mol%), 1,8-bis(dimethylamino)naphthalene (proton sponge, 1.1 eqv), 1-(chloro-1-pyrrolidinylmethylene) pyrrolidinium hexafluorophosphate (PyCIU, 1.4 eqv), and 1-iodobutane (1.1 eqv) in toluene (0.1M) at 120°C (16 hrs reaction time) gave the best results. Continuous venting of the carbon monoxide by-product was required for good yields- presumably due to poisoning of the metal catalyst. PyCIU was an effective deoxychlorinating reagent for in situ activation of the acid.
Kinetic analysis and number of stoichiometric control experiments were carried out and suggested a unique mechanism involving C-P reductive elimination to form the Xantphos phosphonium chloride, which subsequently initiates an unusual halogen exchange by outer sphere nucleophilic substitution (Figure 5). Initial oxidative addition of the acid chloride to the ligated Pd(0) followed by CO release from the palladium complex (A, Figure 5) is postulated. At this point you might expect reductive elimination to give the aryl chloride, but this doesn’t happen. Instead (A) undergoes chemoselective C-P reductive elimination generating (B). It’s suspected that the metal co-ordination sphere contains ligated CO which it is believed could speed up reductive elimination from a more electron deficient metal. Rapid nucleophilic exchange of chloride ion for iodide in the alky halide generates an iodide metal complex that undergoes C-P oxidative addition to form complex (C). Reductive elimination then releases the aryl iodide closing the cycle. In the paper a comment is made that in rare cases, phenyl group scrambling was observed after C-P oxidative addition into the phosphorus-phenyl bond of Xantphos to give iodobenzene. I would be interested in more information on these cases. I guess migration of the phenyl group is less facile than the substituted aryl group. It does however, as they claim, lend support for their proposed mechanism.
A broad range of aryl carboxylic acids were successfully converted to aryl iodides with electron neutral or electron poor acids giving the highest yields, providing much needed orthogonality with existing methodology. Vinyl carboxylic acids could also be converted into the corresponding vinyl iodides- a synthetically useful class of intermediates.
A couple of potentially useful iodination approaches here that certainly add to the state-of the art. Before I go (and to complete my triumvirate) one final paper on iodination- specifically iodination of phenols. A paper by Beifuss et al from 2019 describes a biocatalytic (!) laccase catalysed iodination of p-hydroxyarylcarbonyl- and p-hydroxyarylcarboxylic acid derivatives using KI as iodine source and aerial oxygen as the oxidant. Reported yields are up to 93%.7 We’ll see how a biocatalytic approach gains traction in the wider community. I suspect the reaction is very substrate dependent. But it’s interesting none the less. The introduction to Beifuss’ paper has the longest list of methods for electrophilic iodination you’ve ever seen. A one-stop shop if you want some methods to try!
See you next time.
J
References:
- Large-scale applications of transition metal-catalyzed couplings for the synthesis of pharmaceuticals: J. Dunetz & J. Magano Chem. Rev. 2011, 111, 2177-2250.
- Expanding organofluorine chemical space: the design of chiral fluorinated isosteres enabled by I(I)/I(III) catalysis: R. Gilmour et al, Chem. Sci. 2021, DOI: 10.1039/d1sc02880d.
- Selective C−H iodination of (hetero)arenes: T. Ritter et al, Org. Lett. 2021, https://doi.org/10.1021/acs.orglett.1c01530
- Palladium-catalyzed decarbonylative iodination of aryl carboxylic acids enabled by ligand-assisted halide exchange: B. Morandi et al, Angew. Chem. Int. Ed. 2021, https://doi.org/10.1002/anie.202103269
- Visible-light-photosensitized aryl and alkyl decarboxylative functionalization reactions: F. Glorius et al, Angew. Chem. Int. Ed. 2019, 58, 10514-10520.
- Metathesis-active ligands enable a catalytic functional group metathesis between aroyl chlorides and aryl iodides: B. Morandi et al, Nature Chem. 2018, 10, 1016–1022.
- Efficient and sustainable laccase-catalyzed iodination ofp-substituted phenols using KI as iodine source and aerial O2 as oxidant: U. Beifuss et al, RSC Adv. 2019, 9, 19549-19559.




