
Figure: 1
The idea of using transition metal chemistry in a cellular setting might at first seem counterintuitive- biological conditions are challenging with respect to stability, efficiency and catalyst (not to mention patient) poisoning. Having said that many important intracellular processes are modulated by transition metals complexed in metalloproteins, with functions including storage and transport of proteins, enzyme driven reactions and signal transduction. The most common transition metals found in vivo are Fe, Zn, Cu, Mn, Co and Ni, with iron and zinc being the most significant in terms of concentration.
In recent years the idea of using non-endogenous transition metals to manipulate biological processes or effect selective transformations within a cellular setting have been explored by several academic groups and offers the potential for disease modifying intervention, particularly in cancer therapy.[1] Pd and Ru chemistry have both been successfully utilized in cells- a notable example being Pd0– mediated Suzuki cross coupling in HeLa cells.[2]
The most successful application of a transition metal complex in a clinical setting is the Pt-complex cisplatin, discovered in 1845 and licensed for medical use in 1978 for oncology. It remains a front-line treatment option for 20% of all cancer patients.
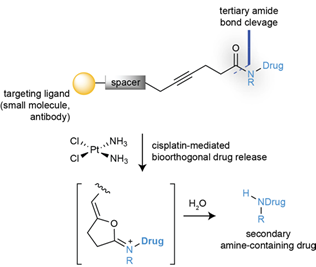
The idea of using a drug as a catalyst to promote a metal-mediated cyclization reaction in a cell is an intriguing one. A pre-print paper just published in ChemRxiv by Bernardes etal (link) describes the bioorthogonal catalytic cleavage of a protected tertiary amides using the drug cisplatin as a catalytic reagent to activate protected analogues of two cytotoxic drugs (5-fluorouracil and monomethyl auristatin E, Figure 1) via cyclization of a carboxylate derivative onto an acetylene (Figure 2). The starting point in their design was the known cyclisation of propargylic acids to enol lactones which was known to be catalysed by platinum anticancer complexes in aqueous media.[3] Initial proof of concept studies were carried out using chloroplatinate complexes (K2PtCl4 / K2PtCl6) in which the metal is present in two different oxidation states. A carbamate (9, X=O) and amide (12, X=CH2) were tested as model substrates, and it was found that the amides cleaved faster and were chemically more stable (Figure 2), most likely due to slower hydrolysis of the stabilized dioxolane intermediate originating from cyclization of the carbamate. The team chose two cancer drugs- the antineoplastic drug monomethyl auristatin (E-MMAE)- a drug suitable for antibody targeted therapies, and 5-fluorouracil (5-FU). Tertiary amides prodrugs with a terminal propargyl group were prepared (Figure 3).
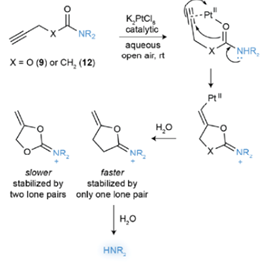
Figure: 2
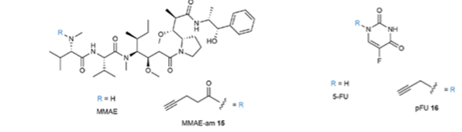
Figure: 3
Incubation of these compounds in HeLa cells with Pt (II) and Pt (III) chloroplatinate complexes at non-toxic metal concentrations resulted in a 2-fold increase in toxicity (by AlamarBlue assay) indicating successful bioorthogonal decaging of the prodrug, albeit with modest results.
To further explore this process in living systems the platinum cleavable caging group was modified with an ADC (antibody drug conjugate). Remarkably platinum mediated decaging was demonstrated to function in a colorectal cancer zebrafish xenograft model using sub-toxic doses of cisplatin to activate the bioorthogonally protected prodrug of 5-fluorouracil, leading to significant tumour reduction in vivo.
This is a fascinating area and I’m sure we will hear more about in vivo application of bioorthogonal metal catalysis over the next few years.
[1] Chem. Commun. 2010, 46, 454
[2] Angew. Chem. Int. Ed. 2017, 56, 6864; Nature Commun. 2014, 3277; Angew. Chem. Int. Ed. 2014, 53, 10536
[3] Nature Chemistry 2011, 3, 239 3 Chem. Commun. 2010, 46, 454




