
One of the things I enjoy most about science, and chemistry in particular, is that even processes that are well understood and used routinely, almost without thinking about them, can be re-invented. The allure of discovering new reactions and being at the forefront of a completely new area of research attracts many academics and students, however I greatly admire those who focus their efforts on fully understanding what we know, the limitations and complexities involved in making reactions useable and scalable and striving for continuous improvement and innovation. I’m reminded of a quote by TS Elliot “We shall not cease from exploration and the end of all our exploring will be to arrive where we started and know the place for the first time”. Ross Denton and his team at the University of Nottingham, UK recently published a paper in Science (2019, 365, 910-914) that addresses some of the intrinsic problems associated with a process first described in 1967- the Mitsunobu reaction.1
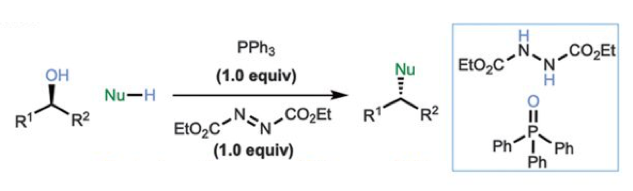
The classic Mitsunobu coupling is a redox process involving reaction of a primary or secondary alcohol with an azodicarboylate (such as diethylaziodicarboxylate, DEAD) and a phosphine (normally triphenylphosphine), followed by reaction of the generated reactive intermediate with a suitable nucleophile (Scheme 1). The alcohol is essentially activated in situ and coupling occurs with the generation of the phosphine oxide (a significant driving force) and the reduced azo-intermediate. The idealised direct SN2 coupling of an alcohol with a nucleophile with concomitant loss of water is kinetically and thermodynamically unfavourable. Both the phosphine and dicarboxylate are used stoichiometrically and generate by-products that drive the poor atom economy synonymous with this methodology. In addition, thermal safety issues with the azo-intermediate often preclude the classical Mitsunobu process from routine scale-up.2
Historically, there have been attempts to develop a catalytic method for this type of transformation. Initial efforts focused on activated alcohols (eg allyl) with transition metals or Lewis or Brosted acid catalysts, use of “borrowing hydrogen” technology and the utilization of cyclopropenone intermediates.3 More recent attempts at redox recycling of the stochiometric reagents used in the Mitsunobu reaction have also been described, however reduction of the phosphine oxide requires a strong, stochiometric reducing agent and/or recycling of the azo-component requires a suitable oxidant compatible with the reaction system.4 That said, Cortney Aldrich has reported a process that is catalytic in phosphine, utilizing 1-phenylphospholane in combination with a phenylsilane (to re-cycle the phosphine oxide).5 He combines this with some work published by Taniguchi on iron–phthalocyanine catalysed oxidation of the hydrazine to azocarboxylate using air as terminal oxidant.6 This work essentially delivered the first catalytic Mitsunobu process (Figure 1).
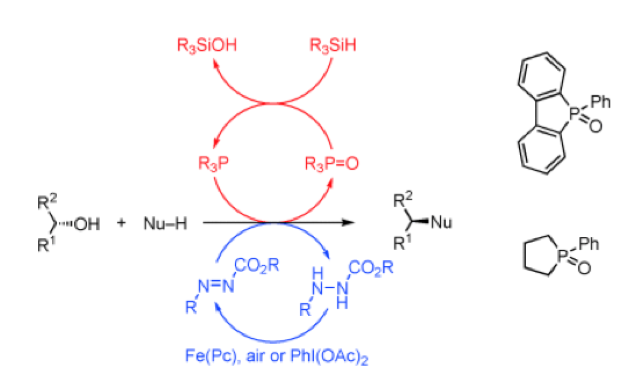
However, reactions based on redox recycling, either catalytic in phosphine, azodicarboxylalte or both were found to be clunky and require stochiometric reagents for reduction/oxidation- which doesn’t really address the atom economy issue. The approach taken by Denton’s team was to target a catalytic process in which the phosphorus atom was invariantly locked in the higher (V) oxidation state. The P(V) organocatalyst they selected (Figure 2) is activated by reaction with an acidic pro-nucleophile and undergoes cyclisation/dehydration to give an oxyphosphonium salt.7 The nature of this intermediate and the equilibrium between the ring open/ring closed forms arising through exchange at the phosphorus center does not affect the cycle, which ultimately transitions to the conventional oxyphosphonium/nucleophile ion pair that goes on to generate the coupled product, regenerating the catalyst. The only by-product in this case is water. In fact the phosphonium salt intermediates(s) are kinetically and thermodynamically unstable with respect to hydrolysis (generating the phosphine oxide) so azeotropic water removal using toluene or xylene solvent is critical to the success of the cycle. Mechanistic studies to underpin the proposed catalytic cycle, including 18O labelling of the alcohol model substrate (1-decanol) and independent synthesis of the putative intermediate phosphonium salts (which were introduced into the cycle to confirm they generated product) proved fruitful.
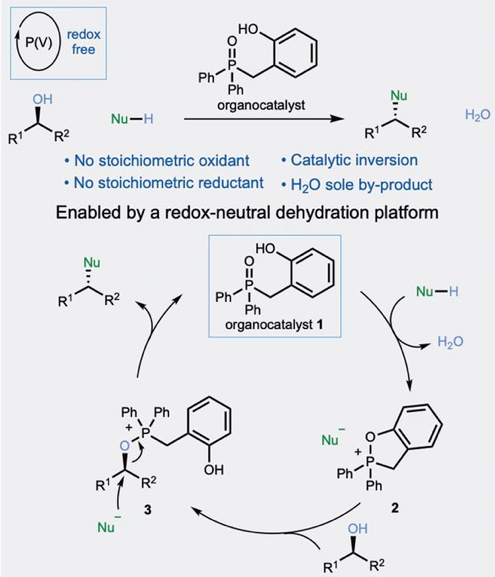
The acidity of the pro-nucleophile was also key for the success of the reaction. Benzoic acid (PKa(H2O) 4.3) was not acidic enough to activate the P=O bond for dehydration. Optimization revealed that dinitrobenzoic acid (PKa(H2O) 1.4) gave good reactivity in the reaction manifold. The optimization was not trivial- increasing acidity led to the formation of elimination products and direct acid-driven coupling gave competing retention of configuration for chiral secondary alcohols. A range of primary and secondary alcohol substrates were exemplified, including some that were substituted with phosphine-sensitive functional groups such as alkyl Br and azide. Chiral secondary alcohols gave the stereochemically inverted ester in high enantiomeric excess.
The method was further extended to include C-N (using an N-N-bis-sulfonamide) and C-S (using thiobenzoic acid) bond coupling, though in the latter example the yield was modest (1-octanol, 35%).
Direct coupling of phenol as a pro-nucleophile was unsuccessful, however, coupling with p-toluenesulfonic acid and subsequent 1-pot reaction with 4-nitrophenol gave an intermediate that was converted to the anti-TB molecule Thiocarlide.8
This is a really beautiful piece of work that has potential application in other phosphorus-mediated transformations. Alcohols are common commodity chemicals and this methodology has the potential to directly convert them to useful building blocks with water as the only by-product.9 Congratulations to Ross and his team.
Footnote:
Many of you may be aware of the recent untimely passing of professor Jonathan Williams, who was head of chemistry at the University of Bath form 2000-2003. His work on transition metal catalysis, including the “borrowing hydrogen”3 methodology referred to above significantly advanced the field of synthetic organic chemistry. He will be much missed by the community. This blog article is dedicated to his memory.
“If I have seen further it is by standing on the shoulders of giants” Isaac Newton, 1675
References:
- Preparation of esters of carboxylic and phosphoric acid via a quaternary phosphonium salts: O. Mitsunobu et al, Bull. Chem. Soc. Jpn. 1967, 40, 2380-2382; ibid 935-939.
- The diisopropyl-azodicarboylate intermediate is frequently used as a safer alternative to diethyl, see development of a pilot-plant-Scale synthesis of an alkylated dihydrobenzothiadiazole S,S-dioxide: incorporation of a late-stage Mitsunobu reaction: T. Connolly et al, Org. Process Res. Dev. 2010, 14, 868-877. For a safer alternative coupling reagent see our earlier blog post: (Cyanomethylene)trimethylphosphorane (CMMP)- a safer reagent for Mitsunobu couplings: Nov. 2018.
- Transition metal catalyzed nucleophilic allylic substitution: activation of allylic alcohols via π-allylic species: C. Bruneau et al ,Chem. Soc. Rev .2012, 41, 4467-4483; Cyclopropenone catalyzed substitution of alcohols with mesylate ion: T. Lambert et al, Org. Lett. 2013, 15, 38-41; The give and take of alcohol activation: J. Williams et al, Science 2010, 329, 635-636.
- The catalytic Mitsunobu reaction: a critical analysis of the current state-of-the-art: R. Denton et al, Org. Biomol. Chem., 2018, 16, 7774-7781.
- Mitsunobu reactions catalytic in phosphine and a fully catalytic system: C. Aldrich et al, Angew. Chem. Int. Ed. 2015, 54, 13041-13044.
- Recyclable Mitsunobu reagents: catalytic Mitsunobu reactions with an iron catalyst and atmospheric oxygen: T. Taniguchi et al, Angew. Chem. Int. Ed. 2013, 52, 4613-4617; Advances and mechanistic insight on the catalytic Mitsunobu reaction using recyclable azo reagents: T. Taniguchi et al, Chem. Sci. 2016, 7, 5148-5159; For use of PhI(OAc)2 as terminal oxidant see P. Toy et al, J. Am. Chem. Soc. 2006, 128, 9636-9637.
- Conditions for preparation of the catalyst are given here. The catalyst is reported to be air and moisture stable. For a review on phosphine organocatalysis see O. Kwon et al, Chem. Rev. 2018, 118, 10049-10293.
- Thiocarlide: (see image below)
- Production of fuels and chemicals from biomass: condensation reactions and beyond: D. Toste et al, Chem. 2016, 1, 32-58.





