
Medicinal chemists love introducing aromatic rings into their pharmacophores. These flat rings make important lipophilic and pi-stacking interactions with proteins and are good synthetic handles for attaching functionality to reach and bind to all the nooks and crannies of an active site. With the advent of cross-coupling chemistry and the simplicity and efficiency of transition metal mediated biaryl couplings (the 2010 chemistry Nobel prize was awarded to Suzuki, Negishi and Heck for developing these reactions), the installation of aromatic rings has become reasonably straightforward. A large number of biaryl compounds populate the screening collections of many pharma companies due to historical uptake of this methodology and generation of huge libraries of compounds via high throughput parallel synthesis.[1] As a result there are a number of pharmaceutically important biaryl molecules used clinically, including anti-inflammatories (NSAID’s), antihypertensives and cholesterol lowering agents. This biaryl motif is considered a privileged structure in drug discovery.[2]
Replicating the cross-coupling chemistry at scale and manufacturing high volumes of materials introduces additional challenges associated with removal of heavy metals, not only from a regulatory and drug safety perspective if the chemistry is used late in the cGMP sequence of API synthesis, but also environmentally in managing waste streams. Much progress has been made in metal scavenging and recycling, however synthetic approaches that avoid the use of metals altogether are particularly appealing.
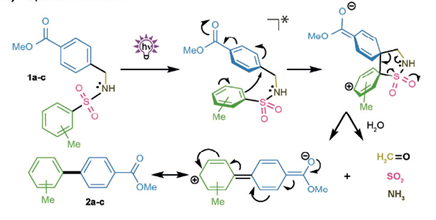
In a clear example of the role of serendipity in reaction discovery, Hertweck and his team at the Leibniz institute have developed a metal-free biaryl coupling by investigating the apparent change in fluorescence of a sulphonamide side-product on a TLC plate (Angew. Chem. Int. Ed. 2018, 57, 1-6). The process involves traceless cleavage of the sulfur-linker with loss of gaseous by-products and is analogous to a biological splicing process (hence the term ‘photosplicing’, see scheme).
Optimization of the reaction revealed methanol and acetonitrile were preferred solvents, with the maximum rate at a wavelength of 233nm confirmed by extensive kinetic studies. The reaction is best performed in a microfluidic device which facilitates a defined irradiation time under continuous flow conditions. However, use of a standard quartz vessel and UV lamp results in only a modest drop in yield. The aryl coupling was exemplified with a range of substituents and gave high regio- and chemoselectivities. Photolabile and photoactive substituents, however, gave very low or no biaryl coupled products. A number of biaryl precursors of clinical molecules were also prepared using this methodology, including Telmisartan, Felbinac and cannabinol.
Mechanistically the reaction is believed to proceed by an ipso-intramolecular cyclization and a formal retro [3+2] cycloaddition, with loss of ammonia, sulfur dioxide and fomaldehyde, all of which were confirmed by analysis of the reaction mixture (Scheme). Formation of these volatile products exert a significant driving force for the forward reaction.
This is an interesting approach and is likely to generate interest in discovery chemistry. The continuous processing aspect coupled with relatively low volumes of waste products is attractive for scale-up and process chemistry.
![]()
[1] Ironically the ease of biaryl synthesis has driven discovery efforts away from 3-dimentional (soluble) structures and into a flat aromatic landscape. The general consensus in medicinal chemistry is that it is not a good recipe for clinical success- see Escape from flatland: increasing saturation as an approach to improving clinical success J. Med. Chem. 2009, 52(21), 6752
[2] Chem .Rev. 2003, 103, 893-930




