
If Nature could re-write her proteinogenic amino acid instruction manual I’m fairly sure that a piperidine ring would feature in there somewhere. Perhaps in the spirit of less is more the pyrrolidine ring won out. Not so for human synthetic engineers- the piperidine ring is ubiquitous in pharmaceuticals, agrochemicals and advanced materials and is the most prevalent saturated N-heterocyclic ring system.1
The modulation of the basicity of the nitrogen atom can have a profound influence on the binding and structural recognition of a piperidine-containing drug and often fluorine substitution is used to this end.2 For example the Merck kinesin spindle protein (KSP) inhibitor shown below (R= 4-piperidine) was a strong P-glycoprotein (P-gp) efflux substrate, however SAR revealed that modulation of the piperidine pKawithin the range 6.5-8.0 by introduction of a fluorine atom in the axial position of the ring resulted in increased basicity better efficacy, and led to identification of the clinical candidate MK-0731 (Figure 1). The equatorial homologue was not pursued (pKa ax 7.6, pKa eq 6.6). In the axial orientation, the C-F bond is antiparallel to the N+-H bond in the protonated substrate resulting in higher relative pKa than the equatorial system.3 This phenomenon has been exploited a number of times in drug discovery.2 Molecules containing basic nitrogen atoms tend to be more promiscuous in vivo and can show cardiovascular tox, driven by interaction with the hERG potassium ion channel, and phospholipidosis.4 Incorporation of fluorine has been used as a strategy to modulate the pKa of an adjacent basic nitrogen atom and in doing so disrupt binding to the the hERG ion channel and increase cardiovascular safety in vivo. To discovery chemists, fluorine and piperidine are a dynamic duo that can have the potential to get them out of deep water. The challenge is bringing them together.
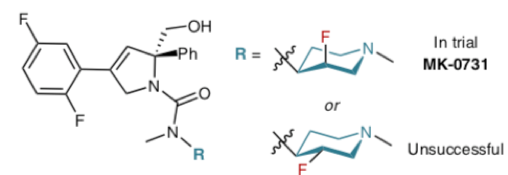
Figure 1: Fluorine modulation of piperidine nitrogen basicity in a Merck KSP inhibitor
The preparation of the fluorinated piperidine substrates, particularly as single isomers, can be difficult synthetically. Having said that several papers have been published recently by both industrial and academic groups that describe synthetic approaches to these molecules.5 Of note is a publication by the Merck process chemistry team (Rahway) that focuses on evaluation of synthetic routes to a syn-fluoro-piperidine pharmacophore required for preclinical preparation of a CGRP (calcitonin gene-related peptide) receptor antagonist targeting migraine (Figure 2).5a
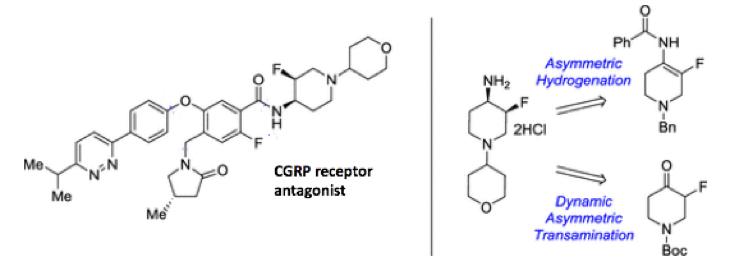
Figure 2: Approaches to a key intermediate for a CGRP antagonist
It’s something of a tradition at Merck to look at both transition-metal mediated and biocatalytic approaches to chiral building blocks and this was no exception. The two approaches the team explored were asymmetric hydrogenation of a fluoroenamide (derived from the corresponding pyridine) and enzymatic dynamic asymmetric transamination of a fluoroketone (Figure 2). Both approaches have the advantage that direct stereoselective fluorination is avoided.
The required asymmetric hydrogenation substrate was prepared by benzoylation of 3-fluoro-4-amino pyridine and activation of the pyridine nucleus for partial reduction by making the benzyl pyridinium salt. Reduction with methanolic NaBH4 gave the fluoro-enamide substrate in a high yielding sequence (Scheme 1). A high throughput screen of commercial phosphine ligands in combination with three metals (Rh, Ru and Ir) was undertaken. Ruthenium and rhodium gave good enantioselectivity (and the required Syn– relative configuration), however C-F bond reduction and formation of des-fluoropiperidine was an issue encountered with both metal systems (iridium promoted extensive C-F bond cleavage). Some neighbouring group effects from the proximal protected nitrogen going on here I would think.6 Ruthenium proved slightly better in this regard and was selected for further investigation.
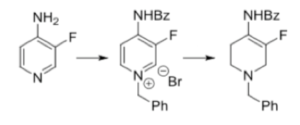
Scheme 1: Partial pyridine reduction to the fluoro-enamide hydrogenation substrate
The active catalyst was prepared from (COD)Ru (Me-allyl)2 and BIPHEP (Scheme 2) in the presence of HBF4 (to prevent substrate inhibition of the catalytic cycle). Substitution of THF (used in screening experiments) for 2-Me-THF (with MeOH co-solvent) was necessary to prevent solvent polymerization and enable substrate dissolution. As many of us have experienced, catalytic reactions can often stall, and in this case conversion frequently plateaued at around 90%. The team traced the problem to fluoride ion formed during formation of the des-fluoro impurity (and I suspect from hydrolysis of the fluoroboric acid). A whopping 2 equivalent of Ti(OiPr)4 was added to sequester fluoride ion! Final conditions utilized 1mol% Ru catalyst and gave the required product in 97% yield (86% ee) with 3% des-fluoro byproduct. SFC purification of the product gave the required optical purity upgrade to 99.8%.
I’m guessing your thinking along the same lines I was upon reading the paper- I hope the enzymatic approach is better. Adding titanium to scavenge fluoride – the proverbial swallowing the spider to catch the fly- together with significant quantities of HBF4 and, in addition to all that, ending up with a relatively low enantioselectivity that needs to be salvaged with a chiral chromatographic separation is not a position you want to find yourself in. Next please.
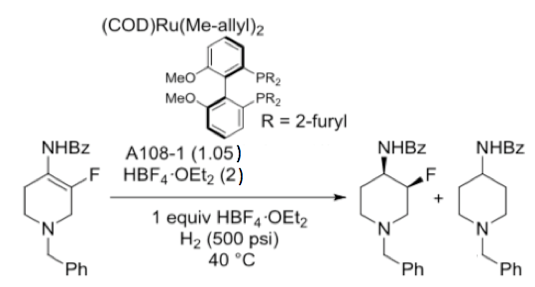
Scheme 2: Asymmetric hydrogenation ((COD)Ru (Me-allyl)2/ Ligand/ HBF4.OEt21:1.05:2)
Before I move on to the enzymatic approach I want to highlight some work published by Frank Glorius, which post-dates the work described above.5c Since conversion of the pyridine to piperidine in the above sequence (Scheme 1) involves partial reduction and de-aromatization to form a hydropyridine followed by hydrogenation of the double bond (both reduction processes), Glorius asked the obvious question- ‘can we do it in a single processing step’? Possible hurdles would likely be catalyst deactivation by Lewis basic heterocycles and hydrodefluorination side reactions. Sound familiar? The team solved these issues by carrying out an initial dearomatization of the pyridine ring using a borane reagent, which also functions to protect the Lewis basic nitrogen, followed by hydrogenation of the diene mixture under milder conditions- a process referred to as DAH (dearomatization–hydrogenation, Figure 3).
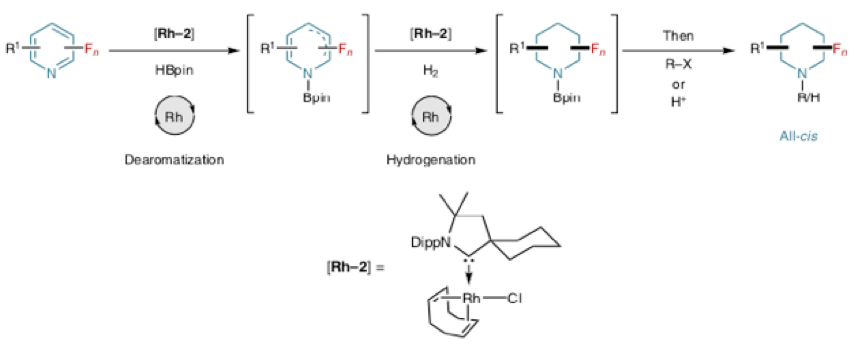
Figure 3: DAH process giving all-cisfluorinated piperidines
Optimization work was undertaken using 3-fluoropyridiene as a model substrate. Stage 1 dearomatization was achieved using a known pyridine reducing system (H-Bpin / [Rh(COD)Cl]2) and hydrogenation of the remaining double bond(s), catalysed by the residual rhodium in the presence of hydrogen gas, gave good chemo selectivity, high diastereoselectivity and respectable yields of piperidine product. A second-generation Rh catalyst (Rh-CAAC, Figure 3) proved optimal.
There are many examples included in the paper, including a protected 3-Fluoro-4-aminopiperidine derivative. Isolation of the (somewhat volatile) products involved derivatisation to the trifluoroacetamide with trifluoroacetic anhydride. Functionalized pyridines gave all-cis products, the fluorine atom preferring an axial orientation in the piperidine ring system. This phenomenon has been seen previously with 3- and 3,5-difluoropiperidine hydrochloride salts and has be rationalized by a favourable dipole interaction (C-F…HN+).7 Hydrogenation of 3-substituted pyridines containing partially fluoro-alkyl groups has also recently been described by Volochnyuk.8
Before I return to the Merck example, I must also mention the glorious work the Glorius group published on direct access to all-cis(multi) fluorinated cycloalkanes, including the unusual, facially polarized, all-cis-1,2,3,4,5,6-hexafluorocyclohexane (Figure 4), from hydrogenation of fluoroarenes using the same rhodium catalyst shown above ([Rh-2]).9a These materials have unique properties, including strongly binding ionic solids such as NaCl (via electrospray ionization and ion trap mass spec),9b and have application in liquid crystal technology.
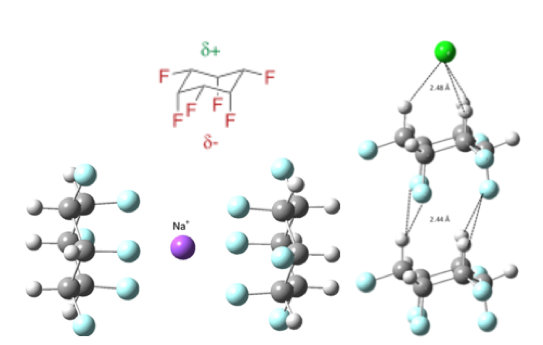
Figure 4: Facially polarized cyclohexane and structure of the complex of Cl– and Na+the dimer of all-cis 1,2,3,4,5,6-hexafluorocyclohexane
Let’s go back to the CGRP inhibitor building block. Biocatalytic technology features heavily in Merck’s approach to green and sustainable processes. Installation of the amino group viaan enzyme catalysed asymmetric transamination, utilizing the 2-fluoroketone substrate (and leveraging the acidity of the C-2 position to enable a dynamic racemization process) was explored as a potential option (Figure 5).
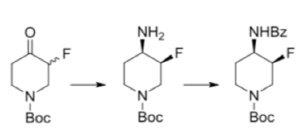
Figure 5: Dynamic kinetic transaminase approach
The fluoro-ketone starting material was made from the Boc-ketone by conversion to the TMS-silyl enol ether and electrophilic fluorination with Selectfluor (91% yield).10 Incidentally a good overview of synthetic approaches to fluorinated pyridines- particularly 3-fluoropyridines- can be found in the intro of a recent paper by Alexander Dilman.11
Screening a commercially available transaminase library (Codexis), using pyridoxal-5’-phosphate (PLP) as cofactor and isopropylamine as amino source at high pH to promote epimerization of the ketone, the viability of the approach was demonstrated. The required syn-isomer, with 2-R configuration was obtained right off the bat. Defluorination byproducts were observed at lower pH on prolonged stirring. Reversibility was avoided by continuously dosing the amine and removing acetone viaa nitrogen sweep. The final optimized process (1 mol% enzyme ATA-3, 1g/L PLP, 0.2M borate at pH 10.5, 1M iPrNH2, 20 vol DMSO, 45°C, 24hrs) gave 66% yield in 15:1 dr (syn) in 96% ee). A further 4 steps (to install the tetrahydropyran ring) completed the synthesis of the intermediate.
Hope you found this article interesting. Rh (CAAC) looks like an interesting catalyst for arene hydrogenation in general-time will tell.12 Please share your thoughts or comments and see you next time.
Written by John Studley, 19th Nov 2019
Scientific Update offer a training course in which we highlight the reasons fluorine is ubiquitous in the pharmaceutical and fine chemical industries, summarize the traditional methods for preparing organofluorine compounds and highlight new developments in the field. For more information contact me at [email protected].
References:
- Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among US FDA approved pharmaceuticals: J. Njardarson et al, J. Med. Chem. 2014, 57, 10257-10274.
- Applications of fluorine in medicinal chemistry: E. Gillis et al, Med. Chem.2015, 58, 8315-8359.
- Kinesin spindle protein (KSP) inhibitors. 9. Discovery of (2S)-4-(2,5-Difluorophenyl)-N-[(3R,4S)- 3-fluoro-1-methylpiperidin-4-yl]-2-(hydroxymethyl)-N-methyl-2-phenyl-2,5-dihydro-1H-pyrrole-1- carboxamide (MK-0731) for the treatment of taxane-refractory cancer: C. Cox et al, J. Med. Chem. 2008, 51, 4239-4252.
- Medicinal chemistry of hERG optimizations: highlights and hang-ups: C. Jamieson et al, J. Med. Chem.2006, 49, 5029-5046.
- a) Synthesis of a CGRP receptor antagonist viaan asymmetric synthesis of 3-fluoro-4-aminopiperidine: E. Philips and B. Xiang et al, J. Org. Chem.2019, 84, 8006-8018; b) Process development for scale-up of a novel 3,5-disubstituted thiazolidine-2,4-dione compound as a potent inhibitor for estrogen-related receptor 1: X. Li et al, Org. Process Res. Dev. 2014, 18, 321-330; c) The formation of all cis-(multi) fluorinated piperidines by a dearomatization- hydrogenation process: F. Glorius et al, Nat. Chem.2019, 11, 264-270.
- See Ref 5b and G. Grunewald et al, J. Med. Chem. 2001, 44, 2849-2856.
- The unexpected diaxial orientation of cis-3,5-difluoropiperidine in water: a potent C-F…NH charge dipole effect: D. Lankin et al, J. Am. Chem. Soc. 2000, 122, 544-545.
- An approach to the synthesis of 3-substituted piperidines bearing partially fluorinated alkyl groups: D. Volochnyuk et al, J. Fluorine Chem. 2019, 224, 61-66.
- a) Hydrogenation of fluoroarenes: Direct access to all-cis-(multi) fluorinated cycloalkanes: F. Glorius et al, Science 2017, 357, 908-912; b) Janus face aspect of all-cis 1,2,3,4,5,6-hexafluorocyclohexane dictates remarkable anion and cation interactions in the gas phase: D. O’Hagan et al, J. Am. Chem. Soc. 2016, 138, 7460-7463.
- Enhancement of oral absorption in selective 5-HT1Dreceptor agonists: fluorinated 3-[3-(Piperidin-1-yl)propyl]indoles: J. Castro et al, J. Med. Chem. 1998, 41, 2667-2670.
- Synthesis of 3-fluoropyridines viaphotoredox-mediated coupling of alpha, alpha-difluoro-beta-iodoketones with silyl enol ethers: A. Dilman et al, J. Org. Chem. 2017, 82, 12967-12974. See also: Pyridines: from lab to production, Ed. E. Scriven, Academic press Oxford, 2013, Chapter 7 (fluorinated pyridines), p 497-516 (W. Dolbier)
- Rh (CAAC)-catalyzed arene hydrogenation: evidence for nanocatalysis and sterically controlled site-selective hydrogenation: R. Bullock et al, ACS Catal. 2018, 8, 8441-8449.




