
Perhaps the most atom economical method for introduction of a carbonyl group into an organic substrate is to use carbon monoxide.1 Millions of tonnes of bulk chemicals are converted to various carbonyl-containing derivatives that are essential commodity products in daily life using this simple C1 unit year on year. Not surprisingly the molecule has a rich history in industrial chemistry (Scheme 1). An early example is the Mond process for purification of nickel as its volatile metal carbonyl. Around the same time Gattermann and Koch developed a process for the formylation of toluene with CO and HCl as the acylating agent under Friedel-Crafts acylation conditions. Arguably the most significant discovery was the water gas shift reaction discovered by Fontana in 1780 in which the equilibrium reaction of CO with water is shifted toward the products carbon dioxide and hydrogen, the latter being a vital component of the Haber-Bosch process for manufacture of ammonia. In the 1920’s Fischer and Tropsch demonstrated reaction of CO with hydrogen over a mixed metal/metal oxide catalyst gave hydrocarbons and in 1938 Roelen developed the ubiquitous hydroformylation process used to instal a formyl (CHO) group and convert olefins to aldehydes. In the 1950’s and 60’s BASF and Monsanto developed industrial processes for the carbonylation of methanol to produce acetic acid. Today catalytic hydroformylation generates annually >12 million tonnes of aldehyde products and intermediates.
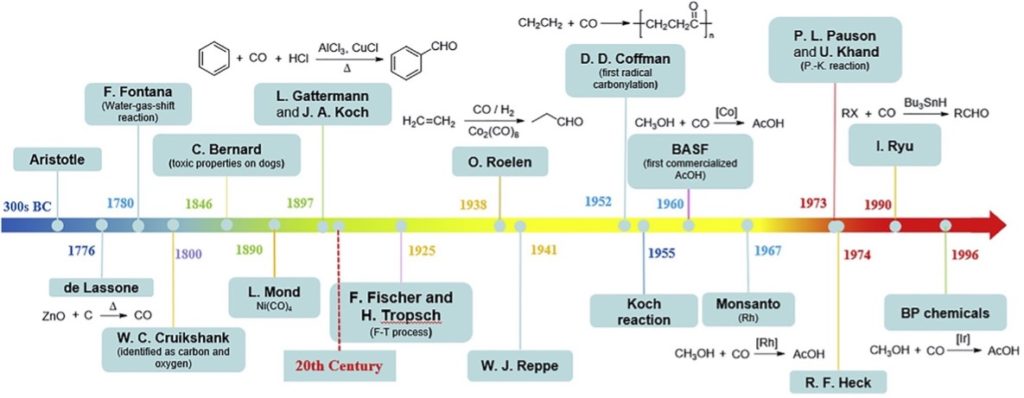
The first transition metal catalysed carbonylations were reported by Reppe in 1953.2 Conversion of acetylene, CO, and water to acrylic acid using Ni(CO)4 as catalyst was operated industrially for a number of years but has now been replaced by other methods.
It wasn’t until the 1970’s that a series of milestone reactions were developed based on palladium-catalysed coupling,3 and it was at this time, in 1974, that the very first palladium-catalysed carbonylation was reported by the Heck group.4 Further developments over the years have focused on methods to avoid handling toxic and highly flammable CO gas, including use of chemical precursors or surrogates for in situ generation of the C1 building block.5
Since those early examples the coupling reaction has been used extensively in both academic and industrial settings, driven by the availability of organohalides, the efficiency of the CO oxidative insertion reaction and the value of the products derived from coupling of a nucleophile and subsequent reductive elimination. Despite the power of this methodology the substrate scope of the classical reaction remains limited. The inhibitory influence of CO coordination to palladium on the oxidative addition requires increased reaction temperatures relative to analogous cross-coupling chemistry and restricts the use of many key substrates such as simple alkyl halides.6 Ligand design has had limited success in mitigating these issues since the electronic requirements for facile oxidative addition and for subsequent reductive elimination are opposing: factors that favour bond cleavage via oxidative addition ultimately inhibit bond formation via reductive elimination. A couple of papers published over the last 12 months describing key advances in the metal catalysed carbonylation reaction are highlighted here.
The first is a paper that directly addresses the oxidative addition/reductive elimination dichotomy. Published in Science by Gerardo Torres and his team, the paper discusses use of visible light to excite palladium and drive both oxidative addition and reductive elimination events with low energy pathways, enabling ambient temperature reactions with a range of substrates ultimately generating acid chlorides, esters, amides, or ketones.7
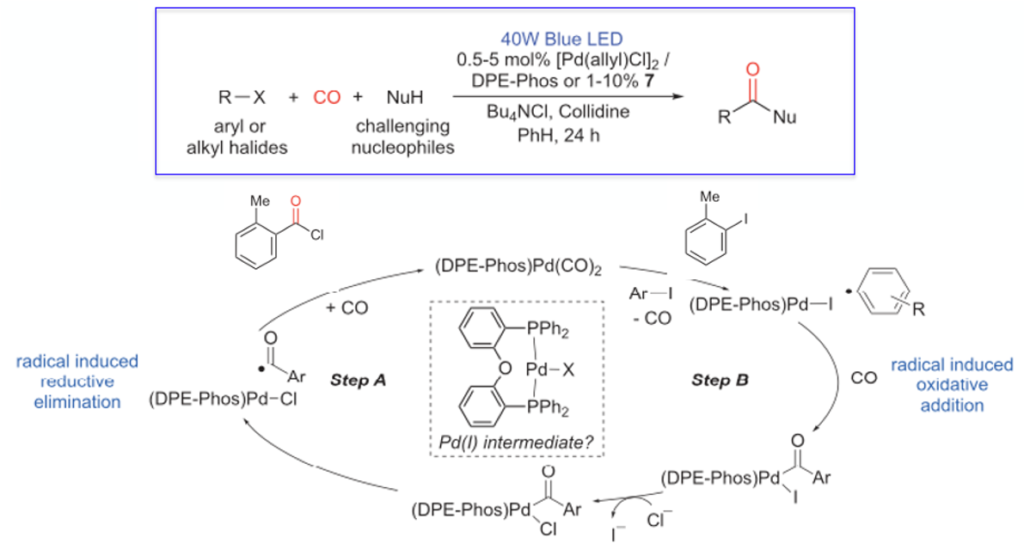
A design concept is highlighted in Scheme 2. The catalyst consist of a Pd(II) precursor and a bidentate DPEphos ligand (bis[(2-diphenylphosphino) phenyl]ether). Application of a low pressure (4 bar) of CO in the presence of a base and chloride source, aryl and alkyl iodides and bromides are converted to acid chlorides. Irradiation of the catalyst in its resting state in the presence of the electrophile results in a SET (single electron transfer) process generating Pd(I) and an aryl or alkyl radical. This low barrier radical species undergoes oxidative insertion of CO followed by halide exchange from iodide to chloride. Similar activation types are not unknown and similar reactions have been described by others, however the reductive elimination step involving a Pd(II) species in combination with Pd(0) photochemistry was previously unreported. What is unique about this reaction is that the catalytic intermediate then undergoes a second irradiation event and an unprecedented radical-induced reductive elimination occurs, most likely through an acyl radical. Since these transformations lead to the formation of acid chlorides, a broad array of nucleophiles can at the same time be incorporated into the chemistry, such as substituted anilines, sterically hindered secondary amines, tertiary alcohols and nucleophilic N-heterocycles.
A couple of very useful applications were also reported in the paper. The first is a direct synthesis of beta-peptide derivatives avoiding the use of multi-step synthetic approaches and the second is trapping the acid chloride with an electron-rich aromatics in a Friedel-Crafts acylation to generate alkyl-aryl ketones (Figure 2). Both could be useful for generating advanced intermediates for discovery chemistry.
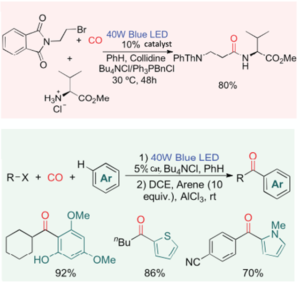
This work should also have wider implications for catalytic processes in general. The ability to change the course of the oxidative addition and reductive elimination pathways within the same catalyst manifold potentially has wider implications and may enable hitherto unknown bond disconnections.
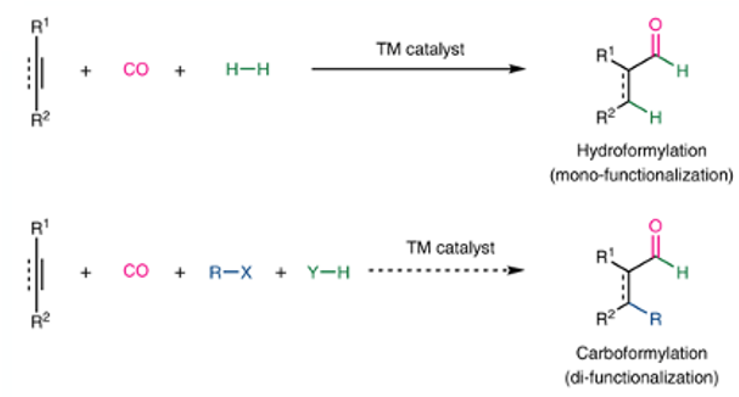
The second, more recent, paper is from Bill Morandi’s group in Zurich focuses on a hitherto virtually unknown extension of the hydroformylation reaction-carboformylation, where a new C-C bond is made in place of a C-H bond (Figure 3).8
Conceptually, based on the value of the resulting products, one would assume this to be a well developed process. However before this report by Morandi only a single publication by Grigg is documented.9 The challenge in moving from a three-component to a four-component system significantly increases the complexity of the system, making for a challenging problem. In short, orchestration of an intermolecular, chemoselective four-component reaction (an electrophile, CO, a hydride source and an unsaturated substrate) that can potentially lead to the formation of more than 10 different products. One way to solve a problem is to simplify it, and this is the approach the team in Zurich adopted. Acid chlorides were identified as both an atom economic source of CO and an electrophilic carbon atom (Scheme 3).
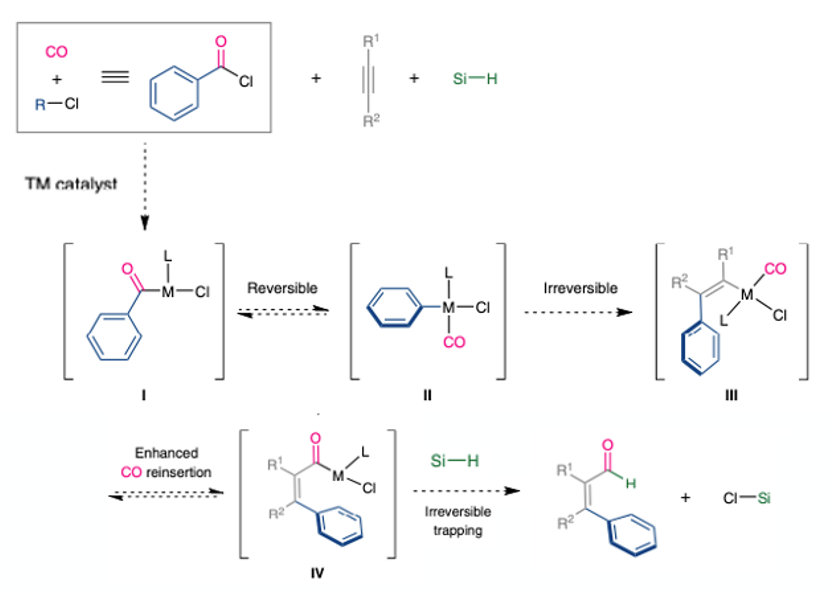
The benefit the acid chloride intermediate brings to the table include introduction of a single molecule of CO (preventing over-insertion with excess of the gas), facile reaction of acid chlorides with low valent transition metals to generate the corresponding aryl–M complex (II, e.g. M=Pd)- that can then add across a triple bond, and generation of an M-Cl bond that can enhance the reductive elimination kinetics allowing the CO insertion to take place. Having climbed mount improbable (and published their results) it’s not surprising that the programmed CO de/reinsertion strategy worked. Using 2.5mol% of Pd with TDMPP or BISBI phosphine ligands and a sterically congested silane reducing agent (iPr3SiH) with 4-octyne as a prototypical acetylene, aldehyde products were obtained (Figure 4).
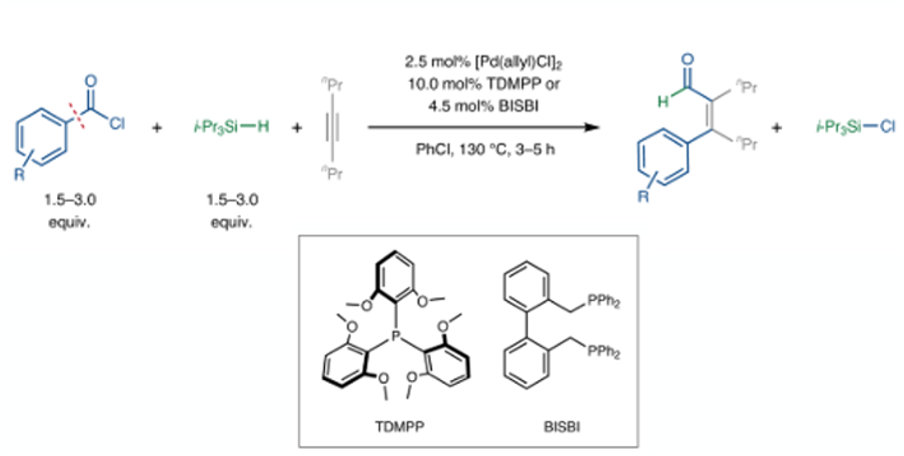
An ortho– structural requirement in the acid chloride appeared to be necessary for good yields in the Pd-catalysed carboformylation and with 4-octyne good yields of α,β-unsaturated aldehydes with excellent (Z)-selectivities are reported. Sterically less hindered aroyl chlorides provided moderate yields accompanied by 5–20% of the corresponding (E)-isomer, suggesting that the ortho-substituents of aroyl chlorides impede E/Z isomerization. The scope with respect to the acetylene was also broad, including silyl acetylene substrates- giving an additional synthetic handle for further elaboration of the products. A reaction run of 5g scale was successful hinting at a possible future in larger scale chemistry. As is common in new methodology papers, incorporation of 13C and deuterium isotopic labels was demonstrated.
The team also looked briefly at incorporation of other nucleophiles (other than hydride) as well as structural variation in the acid chloride (aliphatic v’s aroyol) (Figure 5).
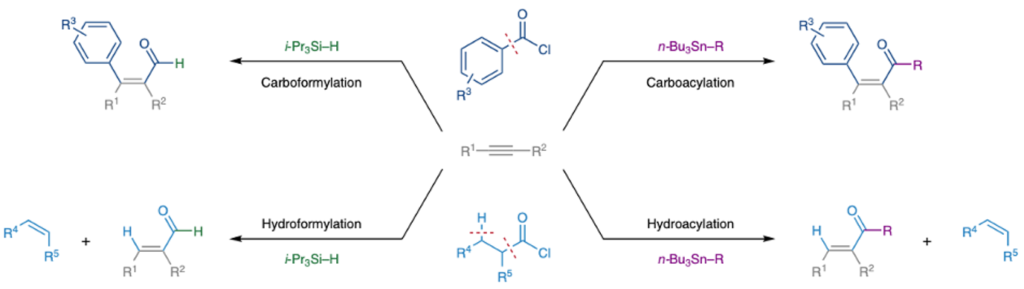
Carboacylation could be carried out using stannane reagents as carbon nucleophiles in place of silanes and use of aliphatic acid chlorides facilitated a mechanistic shift driven by β-H elimination and release of an alkene. The H– Pd(II)–Cl intermediate and CO generated from this fragmentation could then be combined with a hydride or aryl nucleophile to generate the corresponding hydroformylation and hydroacylation reactions (Figure 5). This simple modification greatly increases the diversity of products accessible via this methodology.
Both these papers are a significant step forward in carbonylation chemistry and are likely to be further developed and implemented in coming years.
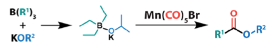
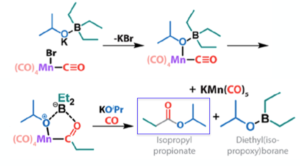
Before I go, another paper on carbonylation- this time using a metal carbonyl complex as a source of CO- has just been published ASAP in Organometallics.10 The focus of the paper is alkoxycarbonylation of organoborane substrates, and a comprehensive review of the palladium-mediated process has just been published by Beller et al.11 In the paper by Pidko et al manganese carbonyl complexes and organo(alkoxy)borate salts react to form an ester product containing a new C-C bond (Figure 6). The organo(alkoxy)borate salts are prepared by direct stoichiometric reaction of organoboranes and alkoxides and are telescoped into the carbonylation step. Aromatic and aliphatic esters are exemplified, including tert-butyl esters. The proposed mechanism is based on reactivity studies, DFT calculation and boron NMR studies (Figure 7).
References:
- The chemistry of CO: carbonylation X. F. Wu et al, Chem 2019, 5, 526-552; First-row transition-metal-catalyzed carbonylative transformations of carbon electrophiles: X. Wu et al, Chem. Rev. 2019, 119, 2090-2127.
- Palladium-Catalyzed Reppe Carbonylation: G. Kiss, Rev. 2001, 101, 3435-3456 .
- From noble metal to Nobel prize: palladium-catalyzed coupling reactions as key methods in organic synthesis: Beller et al, Angew. Chem. Int. Ed. 2010, 49, 9047-9050.
- Palladium-catalyzed carboalkoxylation of aryl, benzyl, and vinylic halides. R. Heck, et al, J. Org. Chem. 1974, 39, 3318–3326.
- Carbonylations of alkenes with CO surrogates. Wu et al Angew. Chem. Int. Ed. 2014, 53, 6310–6320; Evolution of carbonylation catalysis: no need for carbon monoxide: Angew. Chem. Int. Ed. 2004, 43, 5580–5588 ; The development and application of two-chamber reactors and carbon monoxide precursors for safe carbonylation reactions: T. Skrydstrup et al, Acc. Chem. Res. 2016, 49, 594–605 .
- Carbonylation reactions of alkyl iodides through the interplay of carbon radicals and Pd catalysts: Wu et al., ACS Catal.2014, 4, 2977–2989
- A dual light-driven palladium catalyst: breaking the barriers in carbonylation chemistry: G. Torres et al, Science 2020, 368, 318-323; P. Kathe et al, ibid 242-243.
- Palladium-catalysed carboformylation of alkynes using acid chlorides as a dual carbon monoxide and carbon source: B. Morandi et al, Nat. Chem. 2021, 13, 123-130.
- Palladium-catalysed cyclisation-carboformylation. Molecular queuing cascades: Grigg et al, J. Chem. Soc. Chem. Commun. 1995, 1135–1136.
- Manganese-mediated C-C bond formation: alkoxycarbonylation of organoboranes: E. Pidko et al, Organometallics 2021,https://dx.doi.org/10.1021/acs.organomet.0c00781
- State-of-the-art palladium-catalysed alkoxycarbonylations: M. Beller et al, Chem. Front., 2021, 8, 799-811.




