
Biocatalysis has the potential to transform how process chemists make molecules, both in terms of improving existing routes to be more sustainable, and to enable shorter new routes via transformations not feasible using chemical methods. A recent review by a team at Pfizer (S.P. France et. al. JACS Au 2023, 3, 715-735) gives an excellent overview of the known large-scale applications of biocatalysis in the pharmaceutical industry. One key point made by the review is that the timeline for enzyme engineering campaigns is often incompatible with the ever-increasing speed with which novel drug candidates are advancing into and through the clinic. There is also a tension between investing early in resource intensive enzyme engineering campaigns and the high rate of failure of drug candidates in early development. Consequently, the risk exists of getting “locked in” to sub-optimal a non-enzymatic route due to concerns of changes in purity profile of the API and having to build a new supply chain. The review does note that certain classes of enzymes, such as ketoreductases (KREDs) and alcohol dehydrogenases (ADHs), are approaching the status of “off-the shelf reagents” due to there being numerous commercial suppliers with libraries of enzymes available at short-notice for scale-up, but that the majority of enzyme applications remain in late-stage development.
I was therefore interested in a recent paper (S. Barber-Zucker et.al. J. Am. Chem. Soc. 2023, 145, 6, 3443-3453) that applied machine learning to accelerate the the enzyme engineering process by making multiple parallel mutations rather than individual stepwise changes. This papers also helped me to see how the recent breakthrough in enzyme folding and structure prediction enabled by deep learning models, such as Alpha Fold, might be applied in the near future to accelerate biocatalyst development and broaden the range of synthetic transformations possible. Ultimately this novel approach could both accelerate and reduce the resource burden required to engineer new or improved enzymes suitable for large-scale synthesis.
The paper looks at using computational design to identify enantiodivergent C-H oxyfunctionalization enzymes to afford chiral secondary alcohols. The current scalable biocatalytic approach to accessing these stereocenters is typically to reduce a ketone with a KRED enzyme, but this is less efficient than being able to simply being able to selectively oxidize one of the two H-atoms on a methylene group to set the stereocenter. Nature is able to do this via many enzymes, such as P450s which account for approximately 75% of all drug metabolism, but unfortunately P450s have proven very difficult to implement for synthesis scale-up due to their need for expensive co-factors and structural complexity. Recently UPO enzymes have emerged from the fungal kingdom as a potentially synthetically scalable alternative to P450s for preparing chiral alcohols (D.T. Monterrey et.al. Current Opinion in Green and Sustainable Chemistry 2023, 41:100786). Unfortunately, UPO’s are typically poorly expressed by traditional gene engineering technologies, so developing their chemistry by preparing libraries of mutants using traditional enzyme evolution library techniques is likely to result in few hits.
Being able to access selectively either enantiomer of a chiral center is also a key requirement of any general catalytic approach. For example, I remember a colleague developing some very nice chiral anion chemistry using sparteine, but unfortunately this gave the wrong enantiomer and the unnatural enantiomer [(+)-sparteine] was simply not available on any meaningful scale, so an entirely different ligand and synthesis had to be invented. Therefore, the goal of this paper was to take a UPO enzyme hit and try to invert the enantioselectivity for 5 standard C-H to C-O reactions. Naively it might sound easy to take an initial hit and develop it to deliver both enantiomers via stepwise enzyme evolution, but is actually difficult because taking incremental single point mutations is likely to lead to a poorer selectivity of the existing enantiomer rather than inverting the enantioselectivity. Indeed, to invert the chirality one can reasonably expect that numerous mutations have to be made simultaneously, but if one were to try to do this by random mutagenesis then the chances of luckily finding the magic combination are extremely low, so the authors apply a more rational approach.
Typically, initial rounds of enzyme engineering for process chemistry focus first on improving the initial hit for the reaction of interest to increase the reaction rate and selectivity, only in later rounds is the enzyme stability a focus to improve tolerance to organic solvents and other conditions necessary for high throughput (see DOI:10.26434/chemrxiv-2023-cbfs1 for an excellent recent example of implementing enzyme evolution this order of from US Merck & Co. scientists). However, the first step in trying to induce enantiodivergence was to try and stabilize the parent UPO enzyme by making multiple stabilizing mutations at the protein surface before working on the active site. The analogy that I think of is that if you are making major changes to a building the first step should be to build a robust scaffold around the structure to stop it falling down during the renovation. The authors have described this approach more fully as a general strategy for enzyme evolution in J. Weinstein et.al. Current Opinion in Structural Biology 2020, 63, p 58-64
Interestingly none of the variants was significantly more stable than the parent (which had already been through 5 rounds of directed evolution and already included a number of stabilizing mutations), so the next stage was to make changes to the active site using a design algorithm called FuncLib that makes multiple parallel mutations to residues in the enzyme pocket while fixing the conformations of the key amino acid residues that bind the substrate and co-factors. In this case each mutant had 4-5 active site mutations. From the top 50 designs the authors selected the 22 mutants with the lowest calculated energy plus 8 handpicked designs based on observations from previous rounds of enzyme evolution. After synthesis and cloning 24 of the 30 enzyme designs were functionally expressed, which represents a very high success rate compared to conventional methods or combinatorial saturation mutagenesis.
The table below shows the results of the screens for enantiodivergence shown as a heat map where -100% ee is represented a dark blue and +100% ee by dark red, and with lighter shading representing a lower ee in the range between 100 (dark) and zero (gray). The bottom row of the table (labeled PaDa-1) shows the results for the parent enzyme. Of particular note is that for all five of the reactions studied at least one of the mutants preferentially provided the opposite enantiomer to the parent.
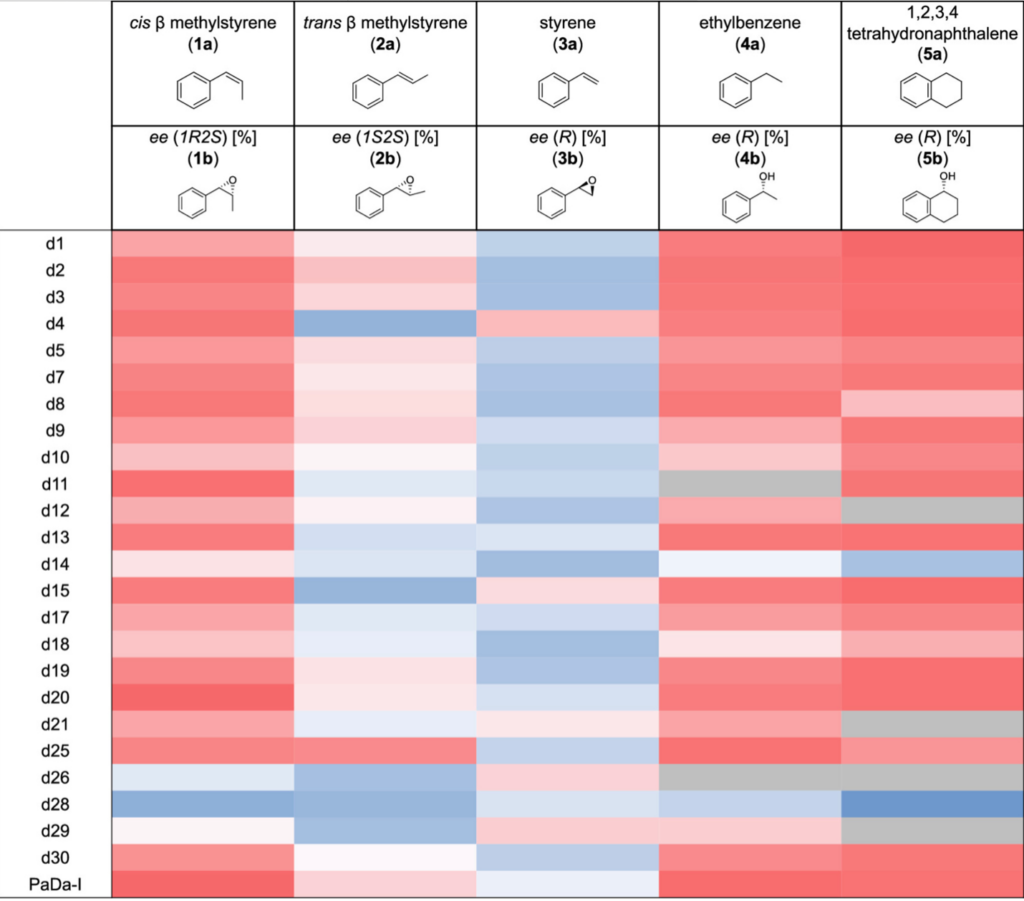
The authors then scaled up and isolated three of the enzymes (d2, d4 and d28) for more detailed characterization, including comparison docking studies that provide a basis for rationalizing the dramatic changes in enantioselectivity. In the case of d28 they show that reversal of enantioselectivity cannot be rationalized by a single point mutation – rather a synergy from multiple mutations is required (which would be highly unlikely to be obtained via conventional stepwise directed evolution).
In the discussion the authors note that the FuncLib algorithm works without transition state modeling, but rather implements stable constellations of interacting amino acids in the catalytic core based on the crystallographic structure of the enzyme. Interestingly they note that the recent high accuracy predictions of enzyme active sites by Alpha Fold models suggests that FuncLib could be operated without even needing a crystallographic structure which would remove one of the most significant bottlenecks that has limited protein engineering to-date. Hopefully, this type of more rational and accelerated approach to enzyme engineering will lead to development of not only UPO enzymes for enantioselective hydroxylation, but also expand and accelerate the whole toolbox of biocatalysis.




