
As with all things in life, necessity is the mother of invention. Synthetic chemistry, as an enabling science, will always rise to the challenge and meet the increasing demand for simpler and more efficient methods to construct fundamentally important structures and functional groups, particularly if these units are conserved privileged pharmacophores imparting biological activity to drugs and agrochemicals. As I’ve pondered a few times on this blog, sometimes science becomes so pre-occupied with progresses we never go back and question what we already know about seemingly simple transformations. A paper published in the fall last year by Ishizuka and Ando on the synthesis of aryl ethers is a good case in point.1
When we think on preparing an aryl ether, thoughts usually turn to transition-metal catalysed cross coupling chemistry such as the copper-mediated Ullman-type reaction or a palladium catalysed version of the Buchwald-Hartwig coupling.2 Aryl halides, typically a bromide or iodide, react with alcohol nucleophiles to give, generally, good conversion to the aryl ethers. Just to really mix things up a paper by David Nicewicz published last year in Organic Letters actually describes cation-radical accelerated nucleophilic aromatic substitution for amination of alkoxyarenes!3
Alternatively, assuming the substrate is suitably activated and able to form stabilising Meisenhemier intermediates,4 direct SNAr type reactions- without the need for transition metals- is possible. Aryl fluorides or chlorides, generally substituted in the ortho– or para– position(s) with a nitro- or cyano- group, react with metal alkoxides (MOR) at high temperatures, usually in a polar aprotic solvents such as NMP or sulfolane (Figure 1).5
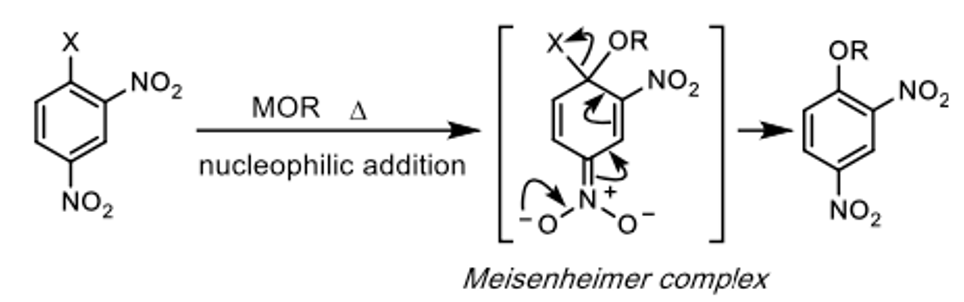
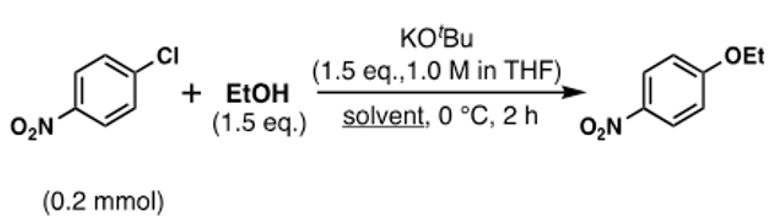
Conceptually, this is the simplest way to make an alkyl-aryl ether- and this is precisely what Ishizuka’s team did. Nothing novel here. Using 4-chloronitrobenzene as a model substrate, sodium ethoxide in DMF (at 0°C, 2hrs) gave poor conversion (24% yield, 49% recovered s.m.) to the aryl ether. Heating would almost certainly have helped, but that would detract from the context of the storey. Pre-mixing potassium tert-butoxide with ethanol in DMF (in situ formation of KOEt) prior to adding the chloroaromatic gave an improved yield (50% yield, 36% recovered s.m.) Again nothing out of the ordinary here. But wait- a dropwise addition of tert-BuONa solution in THF over 5 min to a solution of 4-chloronitrobenzene and EtOH in DMF gave a significant increase in the yield of 1-ethoxy-4-nitrobenzene (86% yield, 13% recovered s.m.). Hmm. And finally, replacing tert-BuONa with tert-BuOK, complete conversion to the aryl ether (97% yield) was observed in <10min (other reactions being reported to take hours). Time to call Mulder and Scully? Let’s think first think about what’s happening here and run a few more experiments.
First a solvent screen- using a commercially available 1M THF solution of KOtBu in a range of polar aprotic solvents revealed that, in addition to DMF, DMAC, DMPU and DMSO were usable albeit giving ether product in lower yields with recovered chloroaromatic (Figure 2). Full consumption of starting material was obtained using DMF (97% yield) and DMSO (74% yield), though the later gave a lower isolated yield due to formation of intractable tars. THF and ethanol as solvents (THF is the vehicle for KOtBu addition and EtOH is the alkoxide precursor) gave no product. Increasing the ratio of THF to DMF resulted in a dose-dependent drop in isolated yield. OK- so something unusual about DMF perhaps, beyond its ability to solvate metal counterions.
Replacement of the chloride with other halogens gave the expected reactivity trend- with fluorine undergoing complete reaction and giving a quantitative yield of aryl ether (conditions as Figure 2). Bromo and iodo both gave lower yields but appeared to be more reactive than expected based on literature precedent.
Is this effect something unique to nitro? Unlikely, and confirmed by a screen of chlorobenzene derivatives with other electron withdrawing groups at the ortho– and para– positions. Ortho– and para– cyano were both highly effective, whereas an ortho- CF3 group was not effective at activating the chloride. 2-chloropyridines were also ineffective, however 2- or 4- chloroquinolines gave quantitative conversion to the corresponding ethers.
A matrix of experiments designed to look at the scope of the alcohol component was carried out using 4-nitro and 4-cyano chlorobenzene as substrate. Primary alcohols worked well at 0°C (all >80% yield), however 2-propanol required warming to 25°C and gave a slightly reduced yield (76%). Other less nucleophilic (e.g. 2,2,2-trifluoroethanol) or hindered alcohols also required warming to 25°C. Increased structural complexity of the alcohol was also successful- examples including menthol, cholesterol and citronellol, all gave ether products in varying yields (see paper).1
Interestingly on several occasions, using less reactive alcohols, 4-nitrophenol was obtained as a major product. All attempts to directly couple water in this system failed, suggesting that in these cases dealklyation was occurring post-coupling.
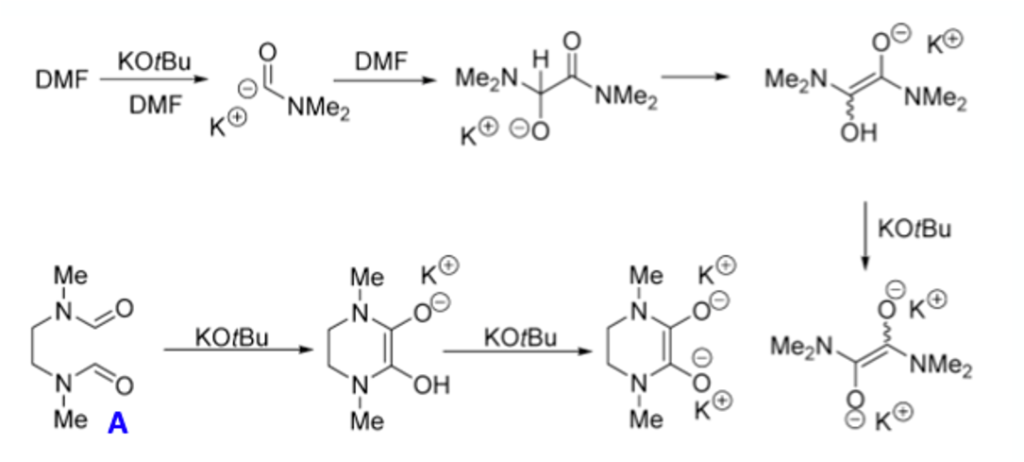
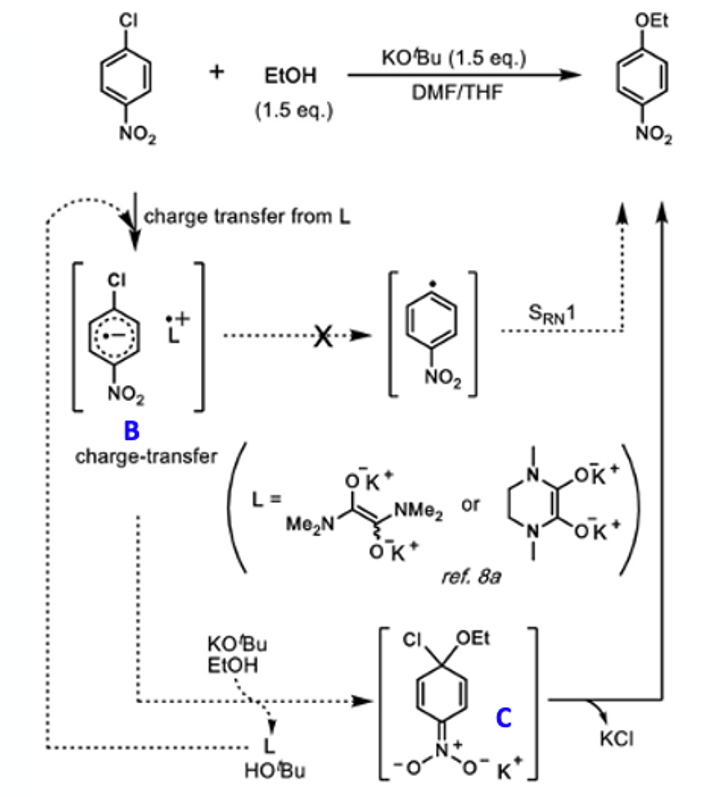
A few control experiments were carried out to enable a working hypothesis to be confirmed. Based on literature evidence, particularly work by Tell Tuttle and John Murphy at Strathclyde university, the team believed that the observed substitution reactions would involve a charge-transfer process via combination of DMF and KOtBu.6
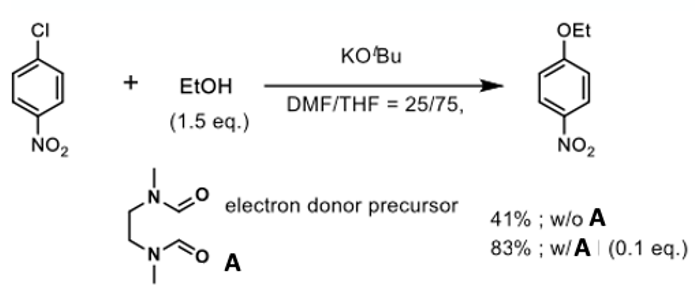
To test this idea they demonstrated that a reaction previously shown to give to give poor yield of aryl ether could be recovered by adding 10 mol% of intermediate (A)- a compound (essentially a DMF dimer) known to promote electron charge transfer processes (Figure 3).6 The high THF ratio in the failed reaction (25:75 DMF:THF) originally gave a yield of 41% which increased to 83% upon addition of (A) (Figure 4). Addition of 1 eqv. TEMPO radical to a previously reported high yielding reaction run under standard conditions gave no reduction in yield. Similarly, an oxygen rich atmosphere appeared to have no effect on the outcome of the reaction (92 v’s 97% yield).
This lack of sensitivity to a radical trap suggested that a mechanism in which the C-X bond dissociated to generate an aryl radical was not likely (Figure 5). The trend observed in varying the halogen leaving group also supports this idea. A more likely mechanism is outlined Figure 5. Reaction of DMF and KOtBu generates a charge transfer complex (L) with the aromatic substrate (B). This species reacts with alkoxide to generate Meisenheimer complex (C), regenerating the ligand complex (L). Loss of KCl then generates the aryl ether product. Further work is required to sure-up all the details behind this proposed pathway.
I hope you agree- an interesting example in which a seemingly simple, well understood reaction can still surprise us. Long may it continue and long may those embarking on their careers in organic synthesis poke and prod at what’s been done before. See you again soon.
References:
- Aryl ether syntheses via aromatic substitution proceeding under mild conditions: S. Ando et al, J. Org. Chem. 2020, 85, 11181−11189.
- Catalytic C-C, C-N and C-O Ullman-type coupling reactions: F.Monnier & M. Taillefer Chem. Int. Ed. 2009, 48, 6954 – 6971; Palladium catalyzed C−O coupling of amino alcohols for the synthesis of aryl ethers: J.Larrow et al, Adv. Synth. Catal. 2020, 362, 430-436.
- Cation radical accelerated nucleophilic aromatic substitution for amination of alkoxyarenes: D. Nicewicz et al, Lett. 2020, 22, 12, 4817–4822.
- New evidence from Jacobsen and co-workers now suggests that these intermediates may only be formed in select cases and that a concerted pathway is more common, see A. Lennox et al, Angew. Chem. Int. Ed. 2018, 57, 14686-14688.
- Concerted nucleophilic aromatic substitution reactions: Murphy et al Angew. Chem. Int. Ed. 2019, 58, 16368-16388.
- KOtBu: A privileged reagent for electron transfer reactions?: J. Murphy et al, J. Chem. Soc. 2016, 138, 7402–7410.




