
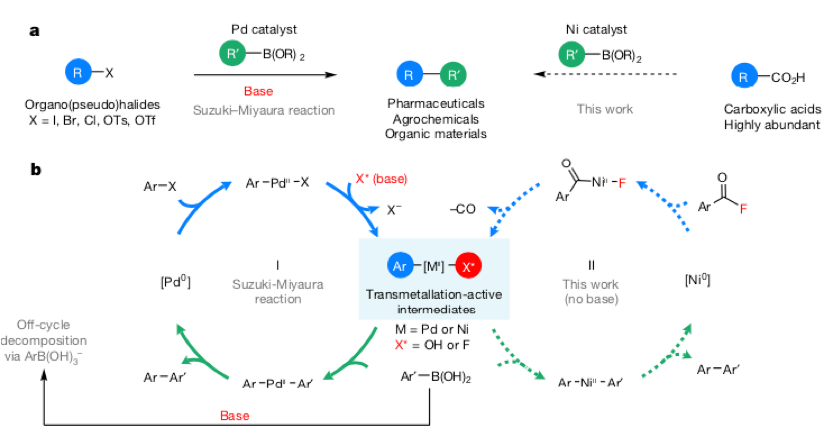
Figure 1: Suzuki-Miyaura Coupling
It’s a reaction that most organic chemists have run at some stage in their careers and is almost certainly in the running for the most widely used C-C bond forming reaction. First reported in 1979, the Suzuki-Miyaura cross-coupling between an organoboron nucleophile and an aryl halide electrophile is still ubiquitous in medicinal- and process chemistry. One might be forgiven for believing that intense academic and industrial scrutiny over the past four decades would have ironed out all the wrinkles in the catalytic cycle and it’s true that the reaction is generally robust and well understood. There are, however, a few inherent problems that remain with some coupling partners. And Murphey’s law says that the substrate you are really interested in always falls into this category! Fear not gentle reader- help may be at hand!
A general requirement of the Suzuki catalytic cycle is that once oxidative addition of palladium has occurred the resulting intermediate [Ar-Pd-X] requires activation by an exogenous base to generating a ‘transmetallation active’ species [Ar-Pd-X*, X=OH or F] to enable facile transmetallation with the boronic acid (Figure 1, cycle 1). In general the addition of base does not cause any ‘off-cycle’ complications, however for a limited subset of boronic acids the organoboronate intermediate can rapidly decompose by multiple pathways including protodeboration, oxidation and homocoupling.1 Competing hydrolytic instability of fluorinated boronic acids in particular can be a challenge.2 Traditional approaches to solving this problem have tended to focus on masking or tuning the boron reagents or designing very active Pd precatalysts. Melanie Sanford’s team at the University of Michigan in Ann Arbor have developed a neat solution to this problem using a combination of a nickel catalyst and acid fluoride electrophile that generates a ‘transmetallation active’ complex via oxidative addition, driven by loss of CO (Figure 1, cycle 2) without the need of a base. Carboxylic acids are readily available and generally inexpensive starting materials and the requisite acid fluorides are generated in situ, making the process very attractive (Nature 2018, 563, 100).
Initial proof of concept studies using stoichiometric conditions (Ni(cod)2, PCy3, benzoyl fluoride) confirmed the oxidative addition occurred rapidly and decarbonylation to the phenyl nickel fluoride complex occurred over 15hrs. Subjecting this ‘transmetallation active’ complex to a boronic acid resulted in full conversion to the biaryl derivative.3 Repeating the reactions using catalytic metal and ligand resulted in formation of a byproduct (the aryl ketone) from competing attack of the boronic acid on the acid fluoride. Modification of the ligand to PPh2Me ameliorated this problem.
In situ generation of the acid fluoride from the free acid was achieved with tetramethylfluoroformamidinium hexafluorophosphate (TFFH) in the presence of proton sponge (1,8-bis(dimethylamino)naphthalene.4
As one might expect there are plenty of examples in the paper demonstrating the wide scope of this process, including coupling of several aryl carboxylate drugs and also organoboron reagents that undergo facile protodeboration, including the known offender- ortho-difluorophenylboronic acid. Revised reaction conditions, replacing Ni(cod)2with a commercially available catalyst Ni(o-tolyl)(PPh2Me)2Cl (10mol%) and CsF (10 mol%) (ostensibly to get around the air sensitivity of the original nickel source) gave comparable results to the original procedure.
This looks like an interesting approach and I look forward to seeing it being applied in challenging cross coupling projects.
References:
- Angew. Chem. Int. Ed. 2013, 52, 7362.
- Eur. J. Inorganic Chem. 2017, 38/39, 4493.
- The corresponding Ph-Ni-Cl and Ph-Ni-Br complexes were inactive in the boron transmetallation step.
- TFFH has been used as a mild deoxyfluorinating agent, see Synlett 2012, 23, 569.




