
Acquisition and processing of analytical HPLC data is often the main bottleneck when performing automated data-rich or high-throughput experimentation. Even though it was over 20 years ago, I well remember spending most of each day running, processing and compiling HPLC data when using an automated reaction platform to run 4 data-rich time course experiments in parallel to prepare a relatively simple amide coupling for the GMP kilo lab. This meant the reactions had to be performed overnight and I had little time each day to think about what was the next best set of experiments to perform.
A recent paper by scientists at MIT and Bayer (Open-Source Chromatographic Data Analysis for Reaction Optimization and Screening https://doi.org/10.26434/chemrxiv-2022-0pv2d) describes an open-source Python project called MOCCA (Multivariate Online Contextual Chromatographic Analysis) which aims to accelerate the HPLC data cycle. The approach analyzes the full time/wavelength data from the DAD array which enables the following advantages over the typical single wavelength data approach:
- Robust peak assignment and quantification
- Peak purity checks
- Deconvolution of overlapping peaks
The other key features of the software is that is uses open-source code so that it is not reliant on the proprietary and relatively fixed software that instrument manufacturers typically supply with their hardware. The authors hope that MOCCA becomes a community project with a significant user base eager to adapt, curate, and further advance the tool. In order to facilitate this community approach the paper includes links to where the authors have published the MOCCA package on the Python Package Index server, made the full Python base code available through GitHub, and provided installation documentation.
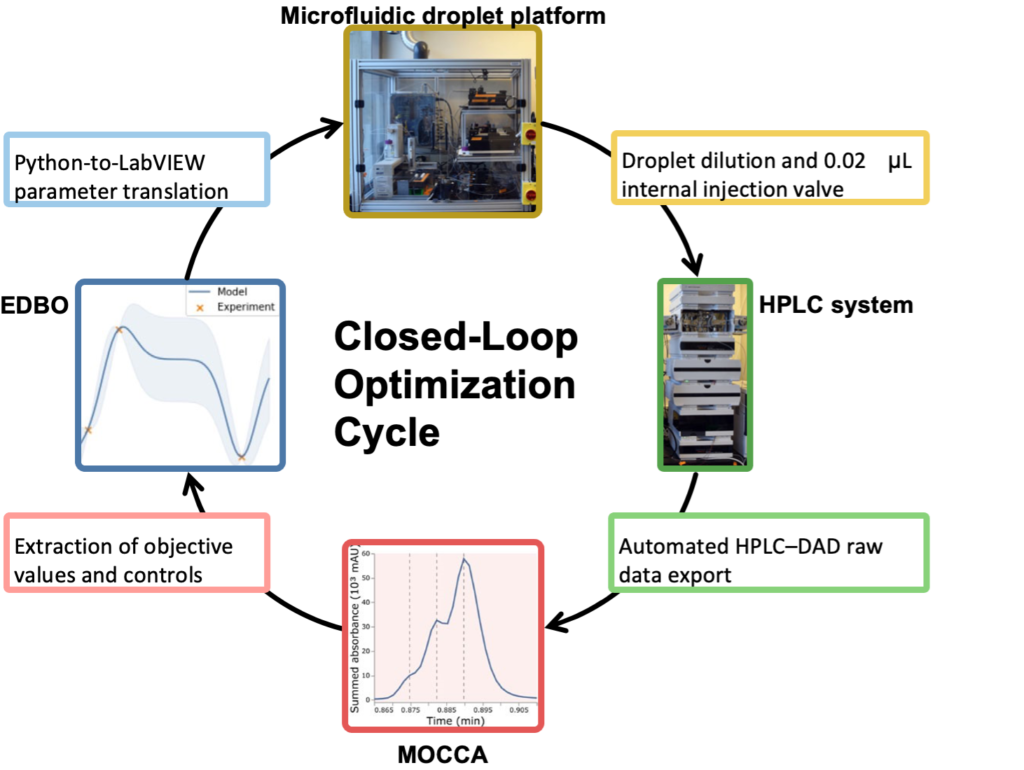
As summarized in the diagram below the proposed analytical workflow starting with HPLC systems controlled by vendor-specific software. The HPLC–DAD raw data are then exported into non-proprietary and open data formats implementing FAIR data principles. After parsing in Python, HPLC–DAD datasets are analysed in context to each other by MOCCA. From the analysis results, structured datasets are generated for data-based decision making by either the chemist or by a computer in an automated closed-loop optimization platform.
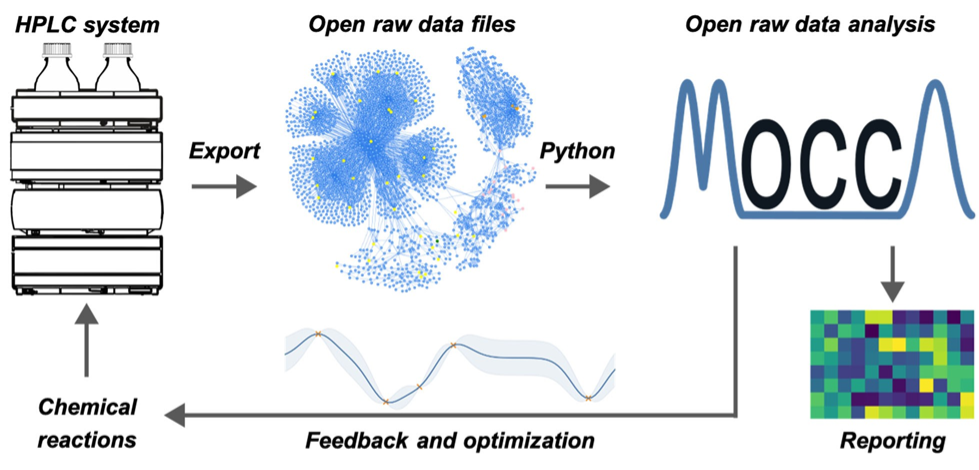
The figures below summarize the three hierarchal levels of data in MOCCA
- The data level includes common features from vendor software, such as baseline correction, peak picking and integration, but also includes less common features, such as peak purity checking and peak deconvolution.
- The information level creates information by aggregating and analysing datasets in context to one another. These processes replace the manual work that a scientist might currently perform to enable peak assignment, peak quantification and retention time correction
- In the knowledge level an interaction tool provides control over certain settings for data analysis and interactive reports on the analysis. These reports include the most crucial information for the user, such as visualization of the chromatograms and peak tables
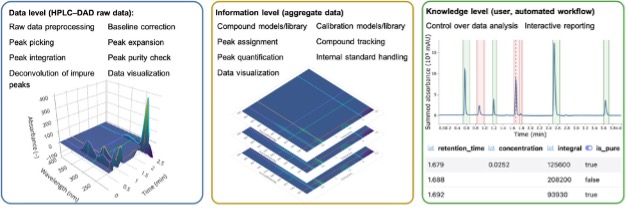
The paper starts by describing how the authors validated the data analysis features of MOCCA by using simulated chromatograms. Naively I would have expected the authors to collect a wide range of experimental HPLC-DAD data but they point out that this would have taken long time and that in any datasets with overlapping peaks there would not be “ground truth” against which the deconvolution results can be compared. The authors identified four result categories where Categories i) and ii) are classified as ‘desired outcomes’ and Categories iii) and iv) are classified as ‘misinterpretations’. The data in the table below show that the deconvolution algorithm is successful in the vast majority of cases, which is sufficient for the type of R&D laboratory setting that this application is designed for. They note that the deconvolution results should be treated with caution for applications in regulated environments or detailed late-stage process development studies
The paper then goes on to give three case studies where the algorithm in applied to real-world optimization studies
The first case study was the well-known Knoevenagel condensation with the twist that two or three aldehydes are simultaneously present in the reaction mixture in order to challenge the peak purity check feature. (Benzaldehyde was treated as the main compound and the other two aldehydes were treated as “unknown” impurities).
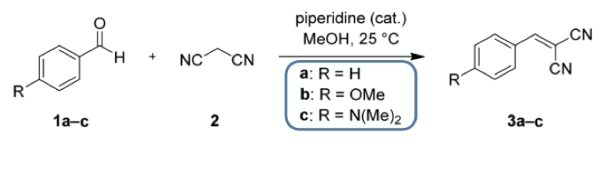
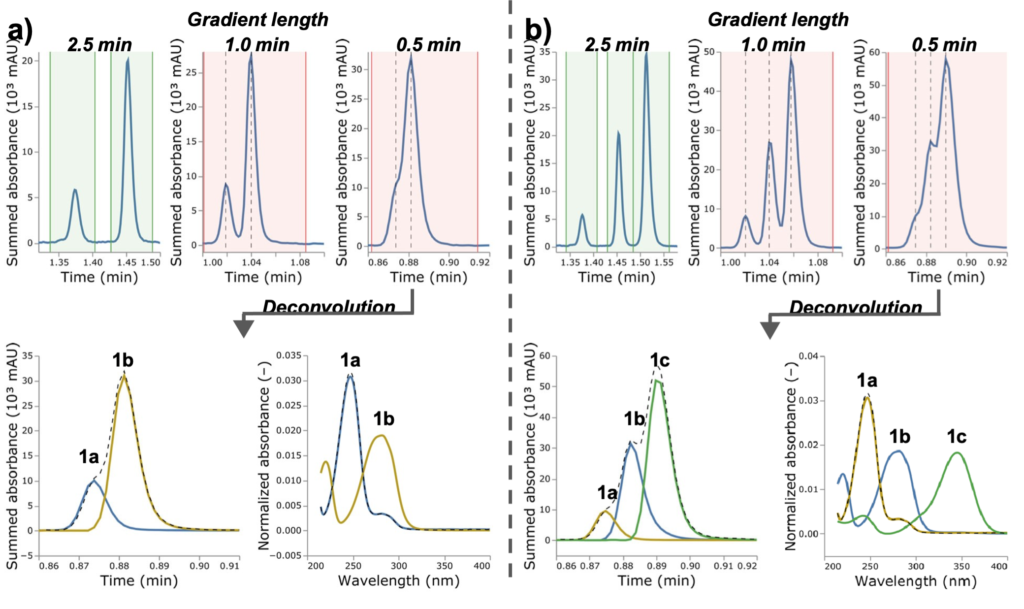
The HPLC analysis was run under five different gradients from 0.5 to 2.5 min. The figure below shows the data from the experiments from two aldehydes on the left and three aldehydes on the right. The top line shows how the peaks overlap as the gradient is shortened to below 2.5 min and the algorithm flags (red) that the peaks are not fully resolved. The lower line shows that even with the heavily overlapping peaks at 0.5 min gradient length the system is able to successfully deconvolute the peaks to give relative intensities that closely align with the ratio obtained for 2.5 min run time. These shorter run times should greatly accelerate the speed of completing an experiment set given the cumulative time savings over multiple samples.
The second application was a closed-loop optimization cycle illustrated below where data analysis by the MOCCA tool are fed into an Experimental Design via Bayesian Optimization (EDBO) Python package that is used to set parameters for experiments in in a microfluidic droplet platform.
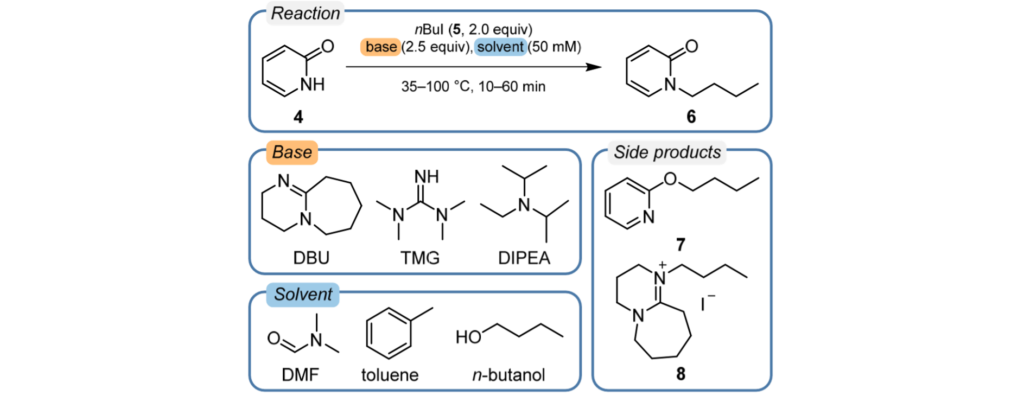
The reaction being explored is the alkylation of 2-pyridone by n-butyl iodide with three bases, three solvents, two reaction temperatures and two run times as summarized below.
The optimization cycle was run with a batch size of one, i.e., the feedback loop was closed after each experiment. As summarized below the best conditions were found to be DBU in toluene for 60 min at 100 °C (Panel a) below).
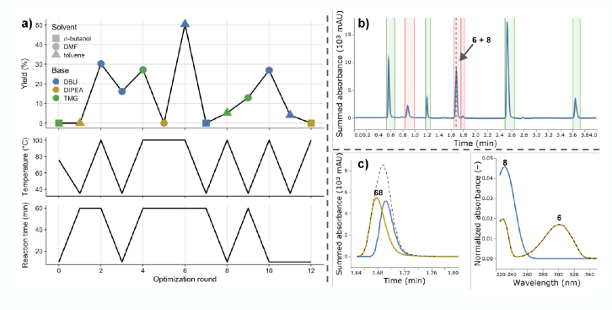
Of particular note is that in the reactions with DBU the HPLC signal of an unexpected side product, butylated DBU (8), started overlapping with the signal of the calibrated product 6 (Panel b) above), whose yield served as the objective value for the optimization. Regardless MOCCA was able to deconvolute this impure peak in an automated fashion (Panel c) above) and to send corrected yields to the design algorithm EDBO and keep the closed-loop cycles running despite this unexpected co-elution.
The final example in the paper is a 96-well plate screening of a range of substrate and catalyst combinations in a modification of the Pd-catalyzed cyanation of aryl chlorides developed by Guimond et. al. This new screen sought to avoid the need to slowly add acetone cyanahydrin by using protected cyanohydrins that would be deprotected in situ.
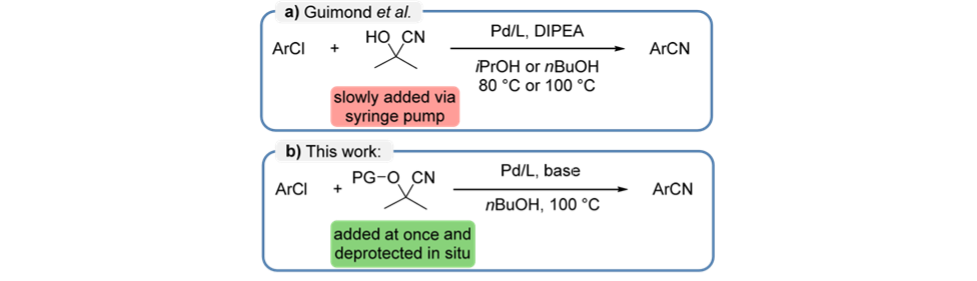
The results summarized below show the results from a well plate exploring 7 protected cyanohydrins against 12 literature precedented combinations of base and ligand. In this case the MOCCA system is being challenged to generate results despite a much wider range of possible unknown impurities.
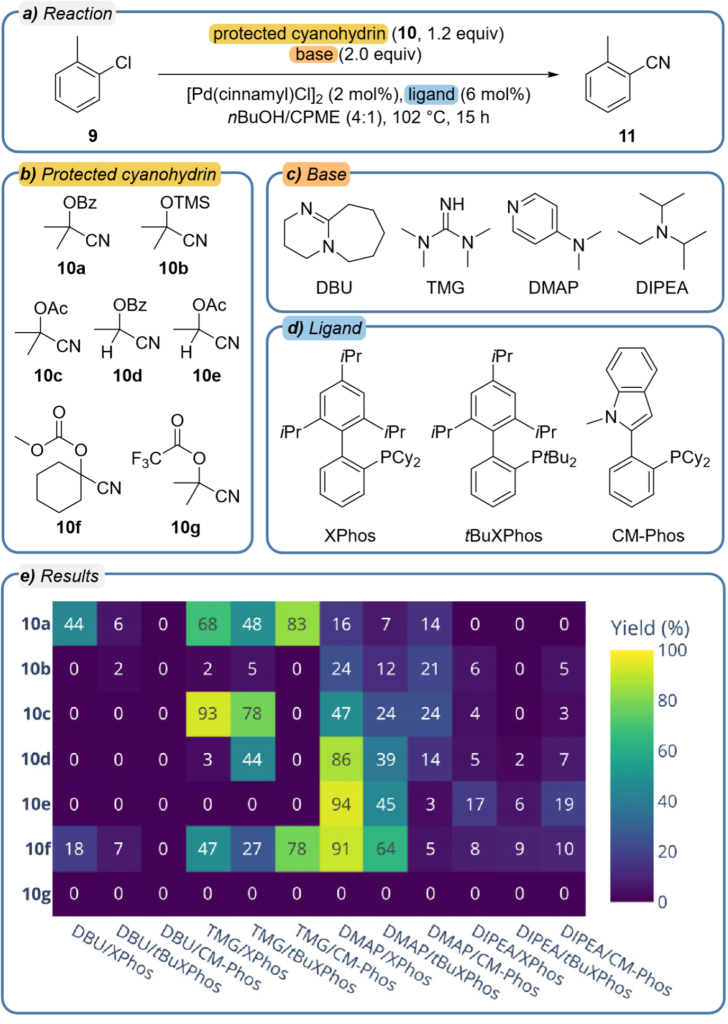
Because the data are exported into Python a wide range of standard toolkits are available to rapidly generate the type of heat map shown above. The authors note that the above data were obtained without the typical HPLC optimization performed before the plate was analyzed to ensure that the method was as short as possible while still resolving all known compounds. When screening combinations there are often a number of unexpected side products produced and these too can co-elute with known signals in the chromatogram, and despite this situation occurring in this screening campaign MOCCA reliably deconvoluted the overlapping peaks.
At the conclusion of the paper the authors foresee the potential addition to MOCCA of future features such as:
- Implementation of a mass spectrometry module to add orthogonal analysis dimensions
- Connection to chemical structure representations
- Linkage to chemical reaction entries in electronic lab notebooks.
These final two features should make synthetic chemistry data and the corresponding analytical data more directly accessible for machine learning in data science applications.




