
One of the most significant technological advancements in synthetic organic chemistry in recent years is the use of photoredox catalysis to generate synthetically useful radical intermediates and promote novel reactivity. It has the potential to enable exploration of chemical space that remains difficult to access using traditional synthetic methodology and too improve the efficiency of historically unaccommodating transformations.1 High throughput screening oflaboratory scale chemistry has been facilitated by innovations in reactor design,2 and several approaches to the scalability paradigm have been reported, notably by engineering teams at Merck and AbbVie.3
Essentially, photoredox catalysis relies on electronic photoexcitation of transition metal complexes or organic dyes with visible light to enable single electron transfer (SET) and generation of synthetically useful organic radical intermediates. The metal complexes employed are mostly precious metal derived, for example Ru(II) polypyridyl or Ir(III) phenylpyridyl (Figure 1a). Their utility relies on the that facile electron promotion to a long-lived triplet-excited state under photo-irradiation is possible and that modification of the coordinating ligands can be used to fine tune the redox potentials of the transition metal complexes.4 Metal free organic dyes such as acridinium derivatives and 4Cz-IPN (Figure 1b),upon irradiation with visible light, are promoted to an excited state that exhibits a long-lived charge separation facilitating their use as single electron Redox catalysts.5
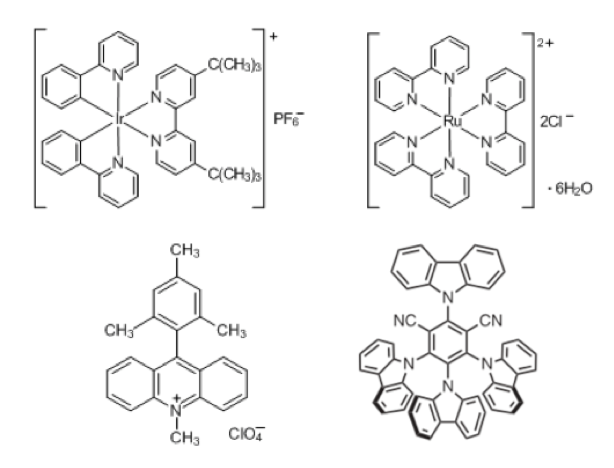
b) Acridinium and 4Cz-IPN organic dye photocatalyst.
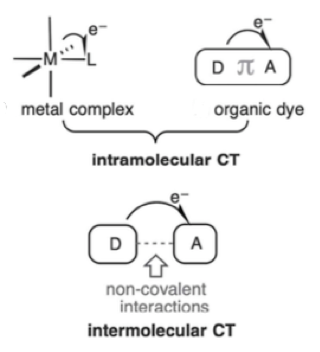
Charge transfer (CT) in an intermolecular sense is also possible through assembly of suitable donor and acceptor molecules via non-covalent interactions. In this case the individual donor and acceptor may not absorb light at the required wavelength individually, but as a self-assembled donor/acceptor complex is able to function as a photoredox catalyst (Figure 2).6
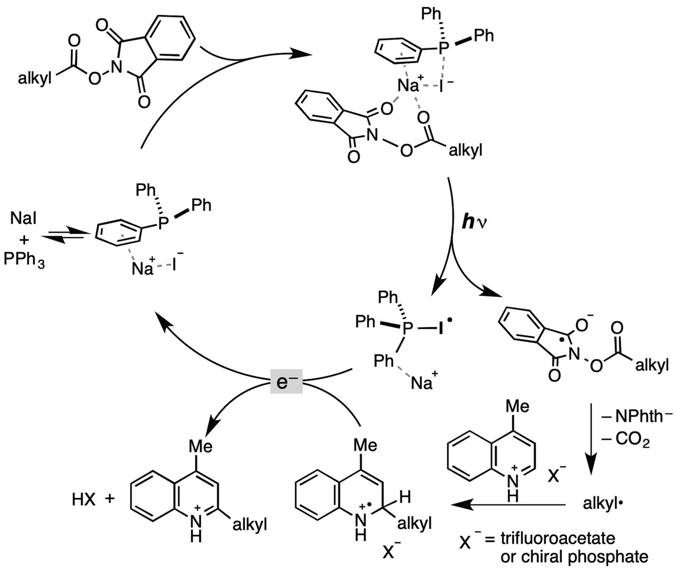
The future industrial application of photoredox catalysisis will be driven in part by the cost of the precious metal catalyst or the synthetic complexity of the organic dye, at least during initial implementation of the technology. A significant step forward in addressing this challenge was recently reported in Science by Fu et al (Science 2019, 363, 1429-1434), who showed that a combination of two inexpensive commodity chemicals (triphenylphosphine and sodium iodide) could promote photoinduced (456 nm, blue LED) intermolecular electron transfer from sodium iodide to an aliphatic redox active ester to induce radical decarboxylation and deliver an alkyl radical for synthetic elaboration (Scheme 1). Coupling of the radical with a silyl enol ether gave good yields of the corresponding alkylated products (20mol% PPh3, 1.5 eqv NaI, MeCN, rt, blue LED). Reaction with N-heteroarenes gave the corresponding Minisci products, again in good yields (20mol% PPh3, 10mol% NaI, 1 eqv TFA, acetone, rt, blue LED).7a
DFT calculations and literature precedence for formation of aryl radicals with NaI in the so called aryl Finkelestein reaction7b suggested that electron transfer from NaI to the activated ester was energetically favourable in the presence of PPh3due to the formation of the PPh3-I.radical, a species that has been detected spectroscopically (Scheme 1).8 Formation of a charge transfer complex is energetically favourable and the energy barrier for electron transfer from iodide to the phthalimide in the presence of PPh3is within 1.5 kcal/mol of the photon energy at 456 nm (61.2 kcal/mol v’s 86.5 kcal/mol without the phosphine). Control experiments without NaI, PPh3or LED irradiation gave negligible conversion, both in the enol ether trapping and the Minisci examples, and sodium seemed to be preferred over lithium or potassium , suggesting complexation with the aryl ring of the phosphine is important (Scheme 1). Catalytic acid was also required to facilitate the Minisci reaction.
Demonstration of the scope of the exemplified chemistry is quite considerable and redox active esters derived from secondary and tertiary aliphatic carboxylic acids as well as a-amino acids, a-hydroxy acids and small peptides are all suitable substrates for Minisci-type reactions. N-heteroarenes including quinolines, isoquinolines and pyridines are described.
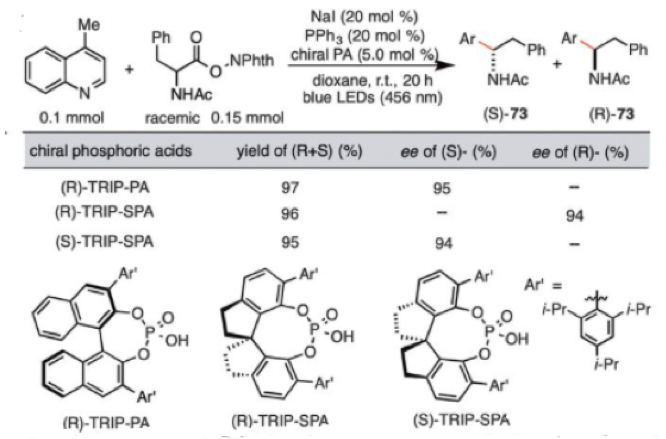
The redox system can also operate synergistically with a chiral Bronsted acid catalyst to promote enantioselective a-aminoalkylation of N-heteroarenes, a reaction recently reported by Phipps et alusing an iridium photoredox catalyst.9 Use of 5mol% of a chiral phosphoric acid, either TRIP-PA or Zhou-type spiral phosphoric acids (TRIP-SPA) gave >95% ee with several redox active esters (Scheme 2).10
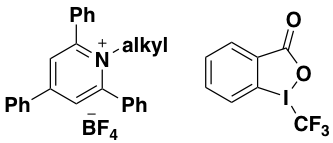
The general applicability of the iodide phosphine redox system was demonstrated by application in the decarboxylative alkenylation of Katritzky’s N-alkylpyridinium salts and trifluoromethylation using Togni’s reagent (Figure 3).11 The tris-4-methoxy- and tris-4-fluoro- phenylphosphines were found to be active in the catalytic process and were advantageous for some reactions.
This paper sets a good precedent for future directions in the design of photoredox catalysts and the almost inevitable use of light energy to effect chemical transformations on an industrial scale.12
By John Studley 23rdJuly 2019
References:
- Photoredox catalysis in organic chemistry: D. MacMillan et al, J. Org. Chem. 2016, 81, 6898-6926; Organic photoredox catalysis: Nicewicz et al, Chem. Rev. 2016, 116, 10075-10166; The merger of transition metal catalysis and photocatalysis: D. MacMillan et al, Nature Reviews Chemistry 2017, 1, 0052.
- A general small-scale reactor to enable standardization and acceleration of photocatalytic reactions: I. Davis et al, ACS Cent. Sci. 2017, 3, 647-653.
- A Laser driven flow chemistry platform for scaling photochemical reactions with visible light: K. Harper et al, ACS Cent. Sci. 2019, 5, 109-115; Merck Centre for Catalysis at Princeton University: http://chemlabs.princeton.edu/macmillan/photoredox/
- Advances in photocatalysis: A micro review of visible light mediated ruthenium and iridium catalysed organic transformations: J. Weaver et al, Org. Process Res. Dev. 2016, 20, 1156-1163; Photoredox catalysts based on earth-abundant metal complexes: N. Robertson et al, Catal. Sci. Technol., 2019, 9, 889-915; Discovery and elucidation of counterion dependence in photoredox catalysis: T. Yoon et al, J. Am. Chem. Soc. 2019, 141, 6385-6391.
- A Toolbox approach to construct broadly applicable metal-free catalysts for photoredox chemistry: deliberate tuning of redox potentials and the importance of halogens in donor-acceptor cyanoarenes: K. Zeitler et al, J. Am. Chem. Soc. 2018, 140, 15353-15365.
- Organic narrowband near-infrared photodetectors based on intermolecular charge-transfer absorption: B. Siegmund et al, Nature Communications 2017, 8, 15421.
- a) Recent advances in Minisci-type reactions: R. Phipps et al, Angew. Chem. Int. Ed. 2019, 10.1002/anie.201900977 b) Photo-induced metal-catalyst free aromatic Finkelstein reaction: C. Li et al, J. Am. Chem. Soc. 2015, 137, 8328-8331.
- Halide ion capture by radicals. Electron spin resonance spectra of R3P·—hal and R2S·—hal σ* radicals: R. Petersen et al, J. Chem. Soc., Faraday Trans. 2, 1979, 75, 210-219.
- Catalytic enantioselective Minisci-type addition to heteroarenes: R. Phipps et al, Science 2018, 360, 419-422.
- Chiral phosphoric acid catalysis: from numbers to insights: S. Wheeler et al, Chem. Soc. Rev. 2018, 47, 1142-1158.
- Photoinduced deaminative borylation of alkylamines: V. Aggerwal et al, J. Am. Chem. Soc. 2018, 140, 10700-10704; Recent progress in the trifluoromethylation of alkenes with Togni’s reagent: C. Zhang ARKIVOC 2014, 453-469.
- Solar photochemical synthesis: from the beginnings of organic photochemistry to the solar manufacturing of commodity chemicals: M. Oelgemoller Chem. Rev. 2016, 17, 9664-9682.




