
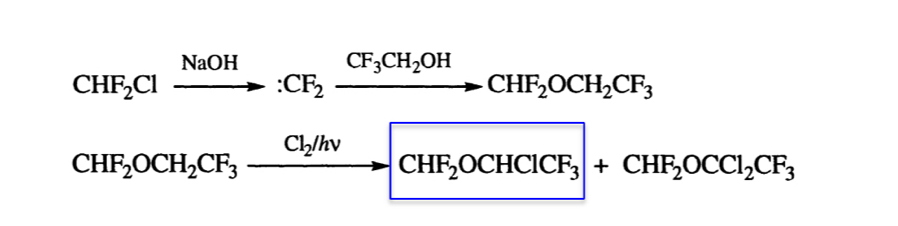
My first experiences of radical chlorination where something of a baptism of fire. Generation of chlorine radicals from chlorine gas under intense UV irradiation in a custom-made photochemical flow reactor was as scary as it sounds to a very young, yet to be trained chemistry technician starting out in his carrier in the chemical industry. The reaction was part of the synthesis of the inhalation anaesthetic Isoflurane and this step was the photochemical chlorination of an intermediate we called FEFME ((2-difluoromethoxy)-1,1,1-trifluoroethane, or trifluoroethyl-difluoromethyl-ether- hence the acronym). Careful control of mixing and flow rates in a gas-phase reaction were critical to prevent over-chlorination, chlorine gas breakthrough and contamination of the precious ethereal product (Scheme 1).1
Chlorine containing molecules constitute a very high proportion of industrial feedstocks and building blocks in the drug, agrochemical and polymer industries. A recent review by Lin estimating that the manufacture of over 50% of industrial chemicals and over 20% of pharmaceutical products require introduction of chlorine at some point in their manufacture.2 Epichlorohydrin, a valuable chloro-epoxide building block is produced on >800 K/Tons per year.
Chlorine-containing pharmaceuticals are frequently a part of the annual cohort of regulatory approved NCE’s reaching patients, with over 250 FDA approved drugs currently on the market for a wide range of disease targets.3 Halogen bonding in biomolecular systems is still a very active area of research in drug discovery, both in the lab and in silico.4
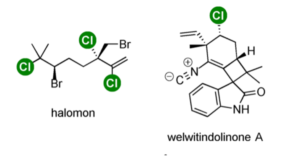
There are, of course, also a vast number of organochlorine (and organobromine) compounds in Nature, some with interesting structural architecture, for example halomon, a polyhalogenated monoterpene first isolated from marine red algae, and welwitindolinone A, an oxindole-containing alkaloid isolated from blue-green algae (Figure 1). These inherently complex architectures being adorned by halogenase enzymes .5
Halogenated materials (organobromine and organochlorine) have had their controversies in the past. Perhaps the most amusing acknowledgement I’ve ever read in a paper was in a review on naturally occurring organobromine compounds by Gordon Gribble.6 I reproduce it in full below:
“The author wishes to thank the environment activist organization Greenpeace for their role in this research. Were it not for their crusade against chlorine and bromine and their accusation that organobromine (and organochlorine) compounds are inherently unnatural (‘The Product is the Poison—The Case for a Chlorine Phase-Out’, A Greenpeace Report by J. Thornton, 1991), the research in the present article [would not have been pursued]”
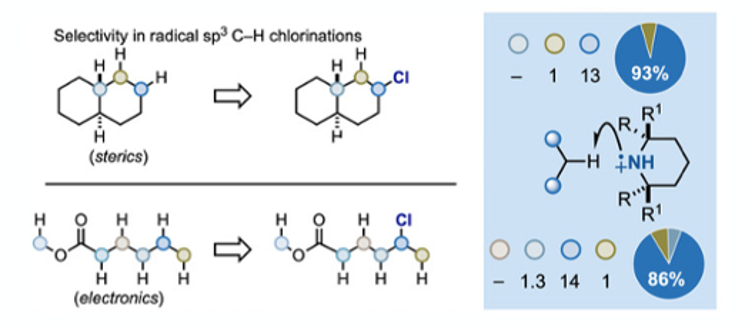
Radical sp3 C-H chlorination is not a new process and is run industrially to make short-chain chlorinated hydrocarbons. However, generation of chlorine radicals and the ensuing chain reaction is difficult to control and somewhat promiscuous in terms of generating mixtures of isomeric (and over-chlorinated) products. Differentiating C-H bonds and gaining selectivity is a significant challenge. A paper published recently by Leonori, Bietti and Ruffoni summaries the challenge nicely by considering two substrates, trans-decalin and methylhexanoate, and their HAT (hydrogen atom transfer) -chlorination product distributions using current state-of the art-methodology.7 The former is largely affected by steric effects and the later electronic factors (Figure 2).
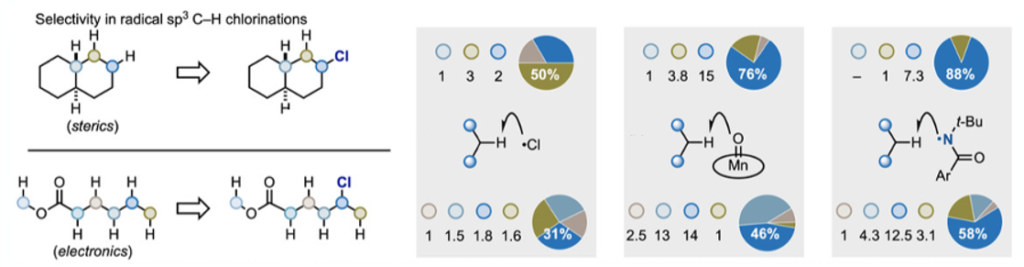
Using the two model substrates, methodology involving direct reaction with chlorine radicals was non-selective from both a steric and electronic perspective (Figure 2, left). Two other methods were investigated. The first a highly fluorinated Mn(III) porphyrin pre-catalyst complex reported by Groves,8 that uses sodium hypochlorite as an oxidant and chlorine source (generating active Mn(V)) (Figure 2, middle). The second was based on work by Greene and later by Alexanian on N-tert-butyl-N-chloroamides amidyl radical precursors (Figure 2, Right).9 Modest improvements over direct radical chlorination but significant room for improvement would be a fair summary here.
Leonori, Bietti and Ruffoni have taken up the challenge and, building on their work on aromatic C-H and olefin amination using photochemically generated aminium radicals, published a report on the ability of protonated N-chloroamines, derived from simple cyclic (commercially available) piperidines, to serve as aminium radical precursors / radical chlorinating agents.7 The mechanistic outline is shown in Figure 3. N-chloroamines (which are powerful electrophiles in the own right) are converted to alkyl radical cationic-open shell-intermediates that had historically been shown by others to perform radical HAT chlorination, albeit under very harsh conditions (using isolated N-chloroamines, high energy UV lambda=254nm, H2SO4 solvent). In this approach amine chlorination, followed by protonation and photochemically induced radical formation begins a chain propagation reaction centred on the aminium radical (highlighted in blue box). The initiation process using blue LED light is believed to occur through adventitious photochemical generation of chlorine radicals that should perform the first (non-selective) HAT event and generate the key radical cation intermediate (chlorine displays a slight absorption in the visible region).10 Presumably acidic N-chlorosuccinimide generates trace amounts of chlorine gas.
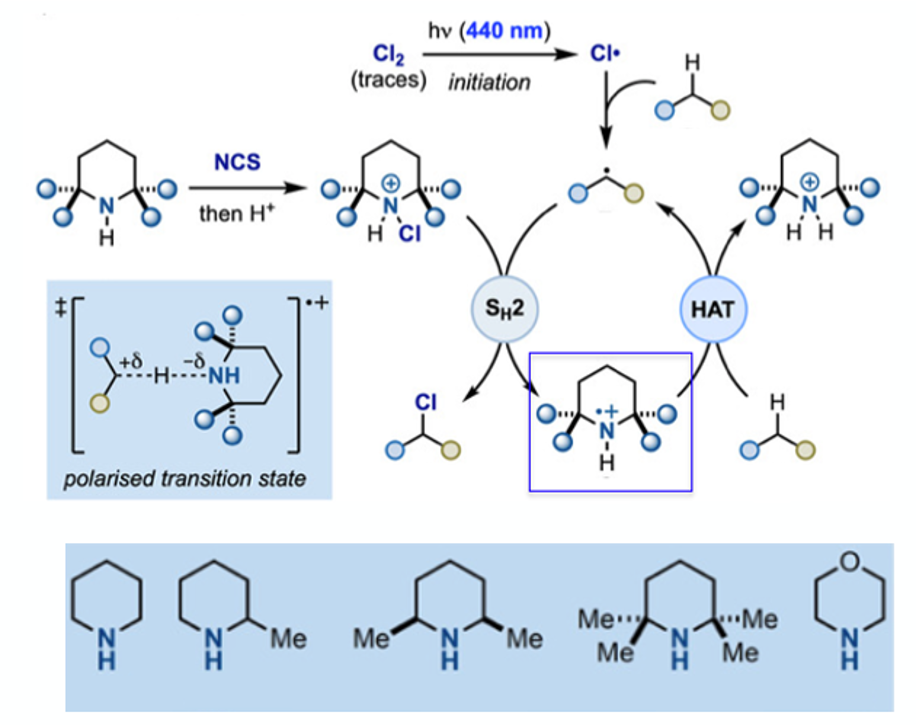
Optimisation of the selectivity of the HAT reaction with respect to aminium radical was initially carried out using trans-decalin as substrate, focusing on steric effects and selective formation of the C-3 chlorinated product (1 eqv. piperidine derivative (Figure 3), 1 eqv. NCS, DCM, r.t., then 1 eqv. Substrate, 3 eqv. HClO4, blue LED’s, 0°C, 2 hrs). 2,2,4,4-tetramethylpiperidine enabled a C-1:C-3 ratio of >13:1 (93% selectivity)- a record for this substrate. A similar approach with methylhexanoate (in which polar effects were likely to dominate) revealed that less hindered amines (piperidine and 2-methylpiperidine), which generate more electrophilc radicals, gave some selectivity for the omega carbon in 84% yield. Isomer ratios were determined by quantitative 13C-NMR analysis of crude reaction mixtures- the volatility of the products proving problematic for other determination methods. A summary of the selectivity data for both substrates is shown in Figure 4.
Mechanistic analysis, particularly around the radical initiation process, was undertaken and revealed that addition of catalytic tetrabutylammonium chloride, known to generate chlorine from N-chloroamines, could re-initiate chain reaction chlorination in failed examples with substrates where chain initiation or propagation were problematic.
A detailed substrate scope is reported in the paper using three different piperidine amines (varying in steric hinderance around the central nitrogen atom, Figure 3) and a range of substrates, with selectivity data presented for each example. The process appears to tolerate a range of functionalities that are often difficult to work with in other radical processes, broadening the scope of the methodology.
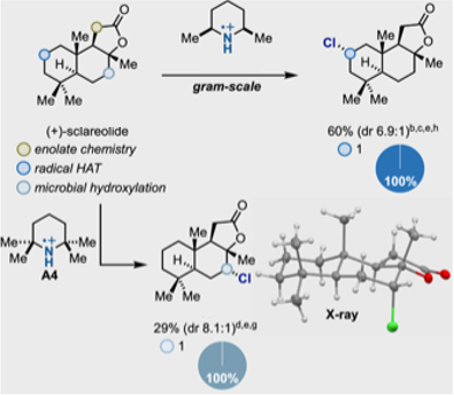
One example I will mention is the sesquiterpene (+)-sclareolide- a benchmark substrate often evaluated in C-H functionalization methodologies (Figure 5). This molecule can be elaborated at C-11 using enolate chemistry, or at C-2 by HAT approaches. Using the optimised conditions described in the paper with 2,6-diemthylpiperdine, selective chlorination at C-2 was obtained in 6.9:1 d.r. on gram scale. Increasing the steric hinderance of the piperidine (2,2,6,6-tetramethyl) switched selectivity to C-7- a position that is difficult to target synthetically. This suggests that reagent-dictated site selectivity for complex substrates might be possible by screening aminium radicals (given the wide range of potential radical amine radical cation precursors available) – a promising approach for increasing access to diverse chemical space for discovery chemistry.
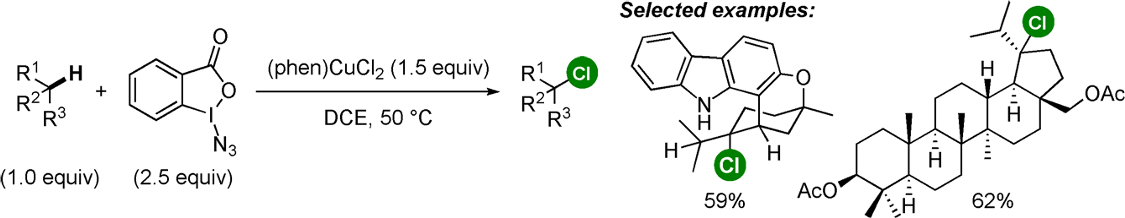
Finally, I should also mention a second paper also published in Angewandte Chemie by Hartwig et al.11 The paper describes highly selective (late stage) chlorination of tertiary and benzylic sp3 C-H bonds using Zhdankin’s azidoiodinane reagent to generate the ortho-iodobenzoyloxy radical for selective C-H abstraction in combination with (phen)CuCl2 to deliver chloride to the resulting alkyl radical (Figure 6). This is similar to earlier work published in 2016 by Chen, who reported use of Zhdankin’s azidoiodinane in combination with lithium chloride and a ruthenium photocatalyst in hexafluoroisopropanol (HFIP) under visible light irradiation.12 However the Hartwig paper avoids photocatalytic radical generation (and strongly oxidising redox catalysts) giving much broader substrate scope. Another method reported recently by Kanai reports chlorination of tertiary and benzylic sp3 C-H bonds using tert-butyl hypochlorite in the presence of a silver catalyst (Ag(phen)2OTf).13 Here promiscuity of the tert-butyloxy radical severely limits the substrate scope. The potential application of these methods, particularly the Hartwig methodology, for late-stage functionalization and diversification of drug-like molecules will, I’m sure, be of interest to medicinal chemistry groups. See you next time.
References:
- Fluorinated inhalation anaesthetics: Organofluorine Chemistry 1994, Ch 25, D. Halpern, Springer Ed. R. Banks; Halogenation using flow chemistry has been reviewed, see: Halogenation of organic compounds using continuous flow and microreactor technology: C. O. Kappa et al, React. Chem. Eng. 2017, 2, 7-19.
- Halogen-mediated conversion of hydrocarbons to commodities: Chem. Rev. 2017, 117, 4182-4247.
- Synthetic approaches and pharmaceutical applications of chloro-containing molecules for drug discovery: A critical review: H. Qin et al, Eur. J. Med. Chem. 2019, 173, 117-153; Beyond C, H, O and N! analysis of the elemental composition of U.S. FDA approved drug architectures: J. Njardarson et al, J. Med. Chem. 2014, 57, 9764-9773.
- Principles and applications of halogen bonding in medicinal chemistry and chemical biology: F. Boeckler et al, J. Med. Chem. 2013, 56, 1363-1388.
- Chlorinated natural products and related halogenases: J. Zeng et al, Israel J. Chem. 2019, 59, 387-402.
- The diversity of naturally occurring organobromine compounds: G. Gribble Chem. Soc. Rev. 1999, 28, 335-346.
- Practical and selective sp3 C-H chlorination via aminium radicals: D. Leonori et al, Angew. Chem. Int. Ed. 2021, 60, 2-10.
- Selective C−H Halogenation with a highly fluorinated manganese porphyrin: J. Groves et al, Angew. Chem. Int. Ed. 2018, 57, 1251-1255.
- Identifying amidyl radicals for intermolecular C–H functionalizations: E. Alexanian et al, Org. Chem. 2019, 84, 12983-12991; ibid Greene et al, 1975, 40, 2192-2196.
- Direct photolysis of N-chloroammonium salts is not possible under blue light irradiation, see S. Fuller et al, J. Chem. Soc. Perkin. Trans. 2, 1981, 545-552.
- Site selective chlorination of C(sp3)-H bonds suitable for late-stage functionalization J. Hartwig et al, Angew. Chem. Int. Ed. 2021, 60, 2-10.
- A visible-light promoted radical reaction system for azidation and halogenation of tertiary aliphatic C-H bonds: Chen et al, Chem. Sci. 2016, 7, 2679– 2683.
- Silver-catalyzed C(sp3)–H chlorination: M. Kanai et al, Org. Lett. 2017, 9, 1430–1433.




