
In June of 1886 (26th to be precise) the French chemist Henri Mossian passed an electric current through a solution of KHF2 in anhydrous HF and became the first person to generate fluorine. Rather him than me. But he did, quite rightly, win a Nobel prize for his efforts in 1905.

Since then, methods to tame this manic material and incorporate fluorine into organic molecules, particularly pharmaceuticals and agrochemicals, has been actively pursued and remains at the forefront of academic and industrial research. You’d be forgiven for thinking that all of the simple methods for installing fluorine into a molecule have been discovered: the low hanging fruit picked- so to speak. However, there are still rich pickings if your prepared to look hard and think carefully about solving the problems inherent in- what appears on paper to be- a relatively straightforward transformation. Many of the existing methods, whilst extremely effective, can be difficult to scale for various reasons (certainly as batch processes) and are therefore limited in scope. I recently came across a few papers on the fluorination and trifluoromethoxylation of aryl and heteroaryl substrates that look both interesting and relatively simple to run. I’ll describe them in more detail here and introduce another, slightly older, paper from 2020 that describes a photoredox cross coupling method to convert your newly constructed (electron rich) aryl fluoride into a more sophisticated aryl amine.
Nucleophilic aromatic fluorination using aryl halides and a fluoride source (the classical “SNAr reaction”) is a tried and tested method of preparing aryl fluorides and has been run on industrial scale for many years. Inorganic salts such as KF, or the more expensive CsF, are utilised at high temperatures in dipolar aprotic solvents. Conditions are harsh- and only the strongest substrates survive (pyridines, activated aromatics etc.). A major complicating factor is the low nucleophilicity of fluoride and its strong, hydrogen-bond driven, association with water. Improvements have been made by the discovery of efficient methods to prepare anhydrous tetraalkylammonium salts- in particular tetramethylammonium fluoride by the Sanford group.1 Remarkably SNAr fluorinations were possible at room temperature, shutting down high-temperature decomposition and widening the substrate scope. That said, the reactions remain extremely sensitive to water- almost to the point that many reactions are on a knife-edge with respect to efficient fluorination and reaction failure. Drying of the ammonium salts can also be a very protracted process and the resulting anhydrous material, not surprisingly, devilishly hygroscopic.2
A team from the Merck process chemistry group recently published a neat extension to this approach- using a dual catalytic system comprising of tetramethylammonium chloride / 18-crown-6 (5 mol%) in combination with caesium fluoride (3 eqv) (Figure 1).3 Aryl triflates and heteroaryl chloride substrates could be converted to the corresponding aryl fluorides in good yields. Importantly the reagents are significantly less hygroscopic than the isolated tetraalkylammonium salts developed by Sanford, and can be manipulated outside of a glove-box.

Not to be outdone by the industrial group, Sanford has very recently published some work on tetramethylammonium fluoride-alcohol adducts (Me4NF·ROH) as fluoride sources for SNAr fluorination (Figure 2).4 The approach is based on earlier work by Kim et al, who demonstrated the use of Bu4NF·(t-BuOH)4 in SN2 fluorination reactions.5 The appeal of these reagents is that they are significantly less hygroscopic that the corresponding tetraalkylammonium fluoride salts and, as with the Merck system, show high reactivity and selectivity for SNAr fluorination without the need for glovebox conditions/exclusion of water.
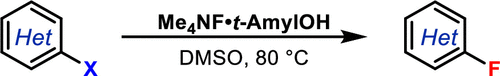
The Me4NF·ROH adducts were prepared by dissolving Me4NF·MeOH in the appropriate alcohol (ROH), removing the volatile components and drying overnight under vacuum.
Since alcohols are potential nucleophiles and can react with activated aryl fluorides, careful tuning of the alcohol was required to arrive at the optimal structure. Perhaps not surprisingly a trend was observed with less etherification observed with increasing alkyl steric bulk. Of the tested adducts Me4NF·t-AmylOH showed the best balance of high fluoride nucleophilicity and low alcohol nucleophilicity. Screening experiments were carried out using 2-chloroquinoline as substrate with 1 eqv Me4NF·ROH in DMSO (the preferred solvent) at 80°C.
The paper exemplifies a large number of substrates, demonstrating wide functional group compatibility and reactivity at the most activated site- independent of the nature of the leaving group. The product yields trended with leaving group in the order NO2 (fluorodenitration)>Cl>Br>I- which is consistent with standard fluorination SNAr chemistry.
A couple of biologically relevant targets were exemplified (Figure 3)- the first a quinazoline core used in the synthesis of a class of antibiotics bearing the same name, and the second 5-chloropicolinate, a structural fragment commonly encountered in agrochemicals.6
This could well come to be the go-to method for SNAr fluorination. Although I have some safety concerns about running reactions at 80°C in DMSO, my feeling is that the academic has the edge on the industrialist- but only time will tell…
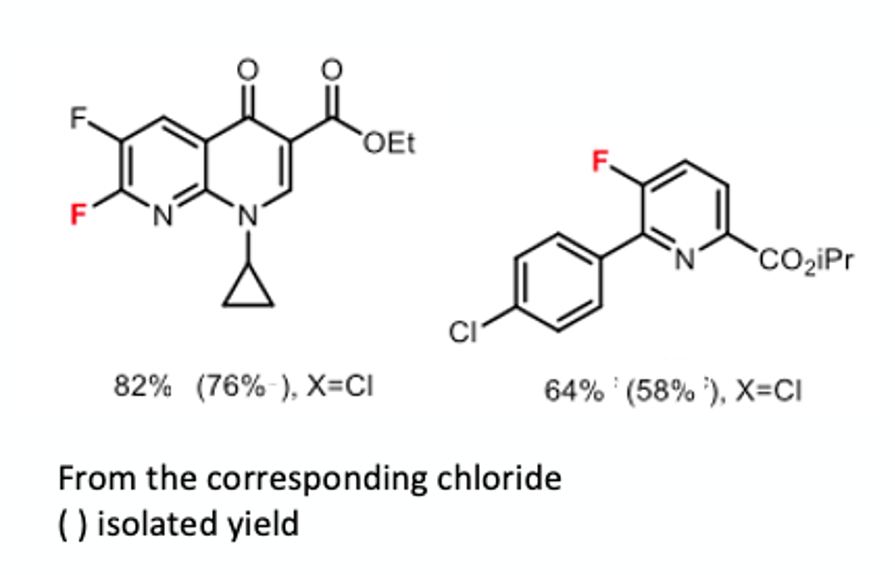
The next paper, again describing work carried out the Sanford group, describes the development of a new reagent for trifluoromethoxylation.7 The -OCF3 group has had wide uptake in the medicinal- and agrochemical worlds as a lipophilic, metabolically stable functional group. The group is usually introduced synthetically via in situ generation of trifluoromethoxide anion from reaction of -OCF3 electrophiles with fluoride or amines (Figure 4). However many of these reagents are expensive or toxic and/or are gases at ambient temperatures.
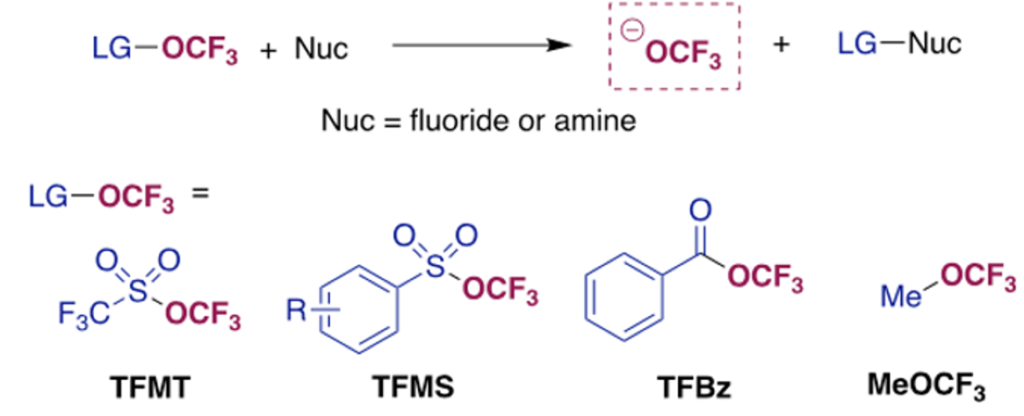
The inventive step in this new offering from Melanie’s team relies on clever leveraging of an inexpensive, commercially available precursor, 2,4-dinitro(trifluoromethoxy)benzene (DNTFB), as an -OCF3 electrophile. Those of you who have read have read this blog in the past might recall that the dinitrobenzene moiety has been used for the formation of pyridinium salts in the Zincke reaction.8 The reasoning here was that reaction of DNTFB with pyridine nucleophiles undergo SNAr with DNTFB to afford isolable trifluoromethoxide salts with a highly delocalized pyridinium cation (Figure 5). This is indeed the case- using THF as solvent the pyridinium salt (with DMAP) can be isolated in 90% yield and 94% purity. Other pyridine derivatives were investigated but were less reactive towards DNTFB.
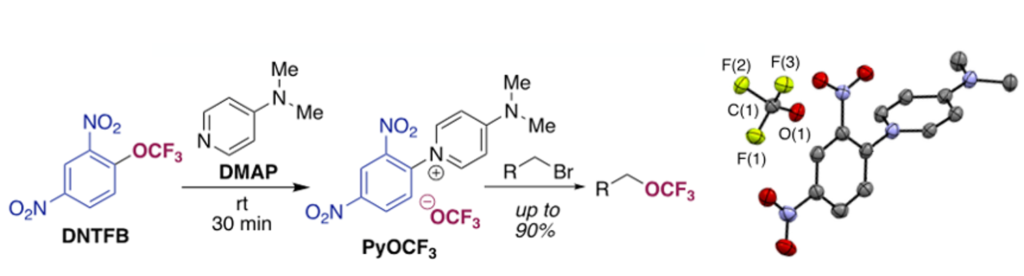
Having powder to store in a bottle is a real advantage and gives much better control of down-stream chemistry. The product (yellow solid) is isolated by direct filtration of the reaction mixture from THF. A significant impurity in the isolated solid was the corresponding 1-fluoro- 2,4-dinitrobenzene, formed by decomposition of trifluoromethoxide to fluorophosgene and fluoride ion followed by SNAr reaction of fluoride with the pyridinium cation.
The PyOCF3 solid is stable upon storage at low temperature (-35°C/1 month) under an inert atmosphere but appeared to be unstable in solution (8 mMol solution in acetonitrile, half-life 30 hrs at room temp).
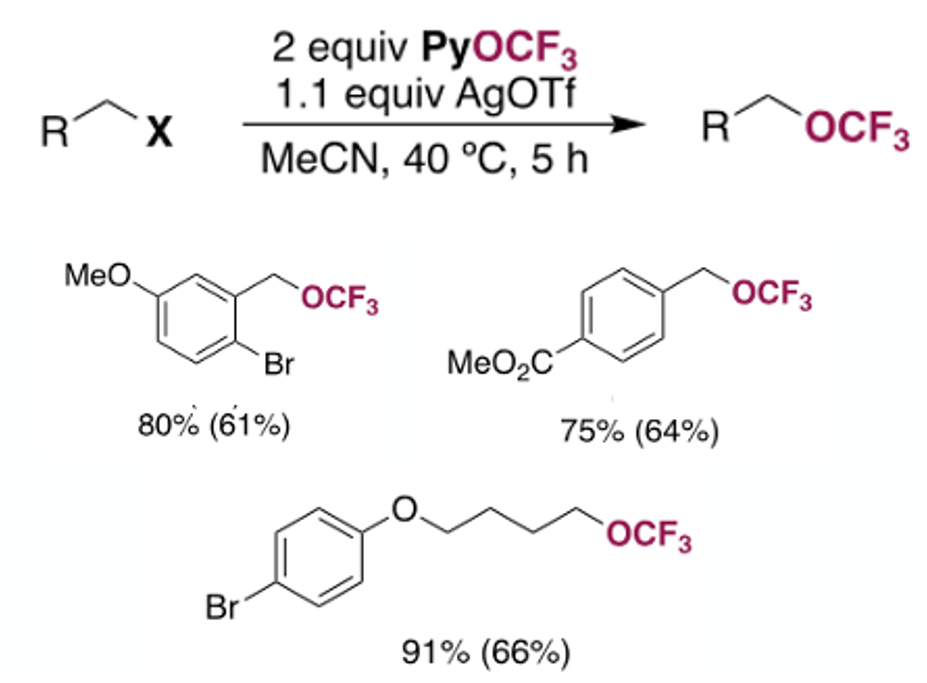
The reagent was used as a nucleophilic trifluoromethoxide source for SN2 reactions of benzyl halides (bromide leaving group) and a small set of alkyl examples (Iodide leaving group). Addition of AgOTf (1.1 eqv) was required to give higher yields of trifluoromethoxy ethers (presumably driven by formation and precipitation of AgBr) (Figure 6). Surprisingly low levels of the corresponding benzyl fluoride were detected- a common impurity arising from decomposition of trifluoromethoxide vide supra.
Sticking with the -OCF3 theme, albeit with a very different mechanistic approach, the group of Matthew Hopkinson have just reported a radical C-H trifluoromethoxylation of (hetero)arenes with the unusual reagent bis(trifluoromethyl)peroxide (BTMP).9 Historically, Nagi, Tongi and Tang have all developed photochemically activated reagent precursors to the .OCF3 radical which undergoes C-H insertion into unactivated aromatics.10 The drawbacks with these systems are the cost, and to some degree the complexity, of the reagents and the poor atom economy associated with stochiometric quantities of organic by-products. Hopkinson’s approach focuses on bis(trifluoromethyl)peroxide as a radical source (Figure 7). At first glance this might appear somewhat fraught with stability (and safety) issues, however perfluorinated peroxides are surprisingly inert. In fact, forcing conditions are required to break the peroxide bond (high temperature or UVA radiation). A recent paper by Riedel et al describes these unusual compounds in more detail.11 BTMP can be made in bulk from CO and F2– or, reassuringly, there are companies out there that can make it for you! Hopkinson solved the problem peroxide bond cleavage problem by using either visible light photoredox or TEMPO catalysis (Figure 7). The paper describes preparation of a wide range of trifluoromethoxylated arenes and, significantly,(trifluoromethoxy)pyridines- which historically have required multi-step synthesis.

In the photocatalytic activation, [Ru(bpy)3](PF6)2 was found to be an efficient single electron reductant (1.5 mol% Ru catalyst, MeCN (0.2M), 16hrs, rt, blue LED’s). The regioselectivity of the photocatalytic trifluoromethoxylation with BTMP mirrored that obtained using existing reagents reflecting the inherent electronic properties of the aromatic substrates.
The TEMPO-mediated process has advantages in that it avoids the use of an expensive metal photocatalyst and is practically more amenable to batch scale-up. Interestingly a complementary regioselectivity to the photocatalytic method was observed with TEMPO- with higher selectivity for the para-(OCF3)-substituted products with each of the aromatic substrate tested.
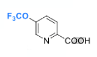
A final section in the paper describes efforts to trifluoromethoxylate substituted pyridines- a reaction that typically requires protracted multi-step synthesis. Application of the TEMPO- mediated process to picolinonitrile gave a mixture of separable regioisomers (albeit in low yield) ,with the 5-trifluoromethoxy derivative being the major isomer. Hydrolysis of the nitrile gave the corresponding acid- a useful synthetic building block (Figure 8).
Bis(trifluoromethyl)peroxide (BTMP) is a potentially useful reagent- once a few fundamental questions regarding its stability and safety have been evaluated appropriate safety engineering teams.
Finally, a paper describing an application of your hard-won aryl fluorides. The classical SNAr reaction, particularly using amine nucleophiles, works well with fluoroaromatics containing electron withdrawing groups that can stable the intermediate Meisenheimer complex. Expanding the scope to electron rich aromatics has historically been difficult to do. Shaolin Zhou published a paper last year, based on some work by Nicewicz on visible-light photocatalytic SNAr reaction of aryl ethers, Zhou’s work utilizing unactivated fluoroarenes with primary aliphatic amines to form aromatic amines (Figure 9).1
Initial experiments with 4-fluoroanisole using tBu2-Mes-Acr+BF4– (5 mol%) as a photocatalyst and valine methyl ester as amine source (3 eqv) with blue LED irradiation (DCE, r.t.) gave 61% yield of the coupled product after 24hrs. Optimization of the ratio of fluor-substrate to amine (1/1.5) and extending the reaction time increased the yield to over 90%.
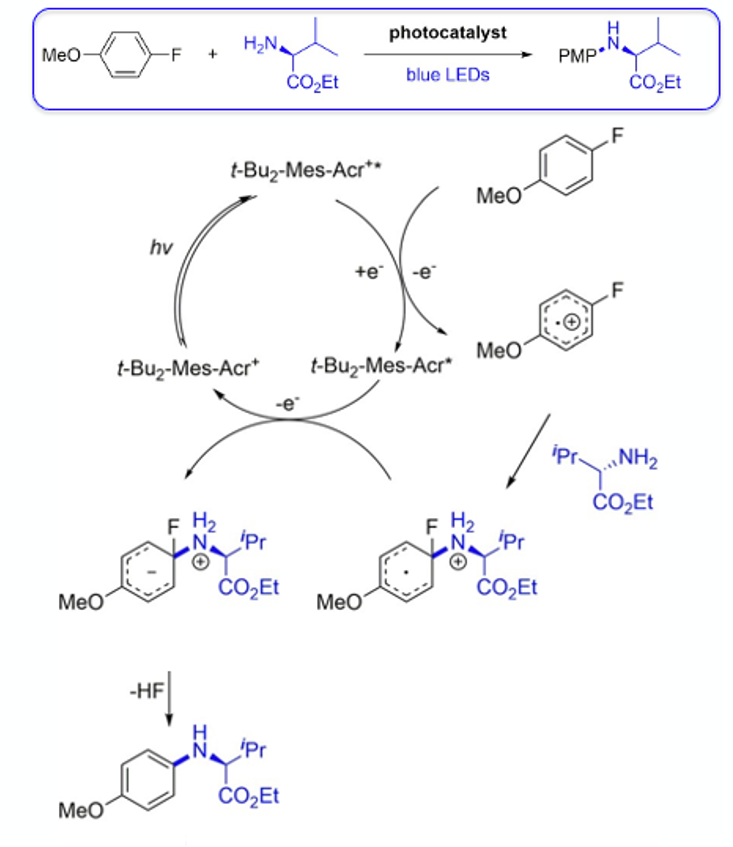
Mechanistically the process is believed to be very similar to other photoredox-mediated nucleophilic substitution reactions (Figure 10). Irradiation of tBu2-Mes-Acr+ forms an excited state photocatalyst, tBu2-Mes-Acr+*, which can be reduced by 4-fluoroanisloe to generate a tBu2-Mes-Acr radical and fluoroarene radical cation. The amine attacks the fluorinated ipso carbon. The radical cation is reduced by the acridiumium radical to regenerate the photocatalyst and produce anionionic intermediate that eliminates HF to give the desired product.
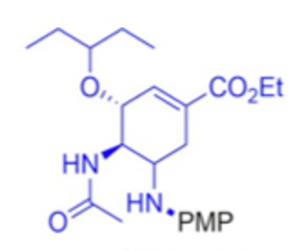
As well as amino acid esters, a-branched and cyclic aliphatic amines were suitable nucleophiles. No reactivity for secondary amines such as pyrrolidine and piperidine was observed. Similar results were obtained with weakly basic amines such as anilines. An encouraging result was obtained with the dipeptidyl peptidase-4 inhibitor Sitagliptin- giving 69% yield of the para-methoxyphenyl (PMP) product (Figure 9, lower scheme). Similarly, the neuramindase inhibitor Oseltamivir gave a 60% yield of the PMP amine.
So there we have it. Some very interesting innovations in fluorine chemistry here, driven by the ever-increasing demand for milder, selective fluorination reactions. Also good to see significant contributions to the state-of-the-art form industrial labs.
Scientific Update run a 2-day organofluorine chemistry training course. If this is something that might be of interest we can run it face to face in-house, online, or, when circumstances permit, face to face as an attended course (hopefully next year). Please contact me if you would like to know more.13
See you next time.
J
References:
- Multiple approaches to the in situ generation of anhydrous tetraalkylammonium fluoride salts for SNAr fluorination reactions: M. Sanford et al, Org. Chem. 2017, 82, 5020–5026.
- Anhydrous tetrabutylammonium fluoride: H. Sun et al, J. Am. Chem. Soc. 2005, 127, 2050–2051.
- Nucleophilic fluorination of heteroaryl chlorides and aryl triflates enabled by cooperative catalysis: C. Hong & A Whittakar et al, J. Org. Chem. 2021, 86, 3999-4006.
- Tetramethylammonium fluoride alcohol adducts for SNAr fluorination: M. Sanford et al, Org. Lett. 2021, 23, 4493−4498.
- Tetrabutylammonium tetra(tert-butyl alcohol)-coordinated fluoride as a facile fluoride source: D. W. Kim et al, Angew.Chem. Int. Ed. 2008, 47, 8404-8406.
- The discovery of Arylex™ active and Rinskor™ active: two novel auxin herbicides; J. Epp et al, Bioorg. Med. Chem. 2016, 24, 362-371.
- Isolable pyridinium trifluoromethoxide salt for nucleophilic trifluoromethoxylation: M. Sanford et al Org. Lett.2021, 23, 13, 5138–5142.
- Pyrylium salts- kryptonite for the Sandmeyer reaction? J. Studley, 2021, https://www.scientificupdate.com/process-chemistry-articles/pyrylium-salts-kryptonite-for-the-sandmeyer-reaction/
- Radical C−H trifluoromethoxylation of (hetero)arenes with bis(trifluoromethyl)peroxide S. Riedel et al, 2021 https://doi.org/10.1002/chem.202101621
- Catalytic C−H trifluoromethoxylation of arenes and heteroarenes: M.Y. Ngai et al, Chem. Int. Ed. 2018, 31, 9645-9649.
- No Fear of perfluorinated peroxides:syntheses and solid-state structures of surprisingly inert perfluoroalkyl peroxides: S. Riedel et al, Angew.Chem. Int. Ed. 2019, 58, 3584-3588.
- Nucleophilic aromatic substitution of unactivated aryl fluorides with primary aliphatic amines by organic photoredox catalysis: S. Zhou et al, Chem. Eur. J. 2020, 26, 14823-14827.
- My contact details: [email protected]




